introduktion
slutförandet av det mänskliga genomprojektet var en relevant milstolpe för medicinsk kunskap, vilket gav den information som var nödvändig för att förstå varje individs unika egenskaper.1 den logiska konsekvensen av denna kunskap skulle vara att kunna tillämpa specifika diagnostiska tester och behandlingar för varje patient baserat på deras individuella genetiska information. Denna nya form av sjukvård kallas personlig medicin.,2 trots de stora framsteg som har lett till kunskapen om det mänskliga genomet har översättningen till diagnostiska och personliga behandlingar varit mindre än förväntat. För närvarande är steg i denna riktning med två stora initiativ: system biologi3 och farmakogenetik.,4 det slutliga målet med dessa initiativ är att utveckla en medicinsk praxis anpassad till egenskaperna hos varje individ som kan förutsäga uppkomsten eller förloppet av en viss sjukdom, tillåta lämpliga förebyggande strategier som skall fastställas och slutligen göra det möjligt för patienten att delta i beslutsfattandet. Detta har kallats P4 medicin.5
cystisk fibros (CF) är fortfarande den vanligaste och dödliga genetiska sjukdomen bland kaukasier., Det sker vid en hastighet av 1 i 2500-6000 nyfödda, beroende på region och etniskt ursprung, och i en del av friska bärare, varierande mellan 1:20 och 37.6 I Spanien, tack vare det gradvisa införandet av neonatal screening program i olika samhällen, en lägre förekomst av CF håller nu på att ses än tidigare beräknat 2009, det vill säga, 1/4430 levande födda i Galicien, 1/4339 i Kastilien-Leon, 1/5376 i Murcia, 1/5840 i Katalonien, och 1/6602 i Balearerna.7 Det uppskattas att det finns 70000 CF-sjuka över hela världen.,8 sjukdomen orsakas av mutationer i genen som kodar för det regulatoriska proteinet cystisk fibros transmembrane conductance regulator (CFTR), en kloridkanal involverad i frisättningen av adenosintrifosfat och reglering av andra jontransportkanaler. Detta protein uttrycks i respiratoriska epitelceller, bukspottkörtel, gallvägar, svettkörtlar och genitourinary system., Dess förändring leder till en abnormitet i jontransport, så att patienter producerar tjock, klibbig slem som täpper till kanalerna i orgeln där den är belägen och så förändringen presenterar multisystemiska effekter som bestämmer det breda utbudet av kliniska manifestationer av CF. Trots stora framsteg i behandlingen av CF som har resulterat i längre överlevnad (nuvarande median uppskattas i 37,5 år) 9 finns det fortfarande en lång väg att gå för att säkerställa att patienter med CF har en kvantitet och livskvalitet som liknar den hos försökspersoner utan sjukdomen., I detta sammanhang är nya behandlingar för att minska sjukligheten och öka överlevnaden nödvändiga.
CF är ett exempel på en sjukdom som är väl positionerad för att dra nytta av personlig medicin. Å ena sidan är det en monogen sjukdom, orsakad av mutationerna i en specifik gen. Enhetens patofysiologi är väl karaktäriserad och de terapeutiska målen är tydliga. Vidare kräver diagnos av sjukdomen genetisk testning för identifiering av sjukdomstypen, så den exakta genetiska defekten bestäms i varje enskilt fall.,För närvarande är två mycket olika tillvägagångssätt inriktade på att korrigera den grundläggande defekten: genterapi, som syftar till att korrigera den genetiska förändringen och molekylterapi, som syftar till att korrigera den funktionella defekten på proteinnivån. Fokus för genterapi är införandet av normala genkopior i luftvägarna hos CF-patienter. Det innebär införandet av en rekombinant viral vektor, vars DNA har extraherats och ersatts av det nya terapeutiska DNA. Den virala vektorn fungerar som ett fordon för att sätta in det nya DNA i målcellen., Olika typer av virus, såsom adenovirus eller lentivirus, har hittills använts. Vidare har icke-virala partiklar, såsom nanopartiklar som kan infoga DNA, också utvecklats.11 resultaten hittills har dock varit dåliga, eftersom varaktigheten av uttrycket av den introducerade genen var kort med båda typerna av vektorer.12 UK Gene Therapy Consortium utvecklar en klinisk fas II-studie för att utvärdera den kliniska effekten av en optimerad PLASMIDISK / liposomal DNA-vektor.13 rekryteringsmålet är 130 patienter och resultaten förväntas 2014 (nct01621867).,14
å andra sidan har terapi som syftar till att återställa funktionen hos CFTR-proteinet varit mer framgångsrik. Under de senaste åren börjar resultaten komma igenom på droger som verkar direkt på CFTR-proteinet. Faktum är att i januari 2012 marknadsfördes det första läkemedlet för att korrigera Gly551Asp-mutationsfel i USA. I följande avsnitt kommer vi att granska tillgänglig information om utvecklingen av personlig medicin för CF och tillgängliga behandlingar som syftar till att korrigera defekten som orsakar sjukdomen på proteinnivå., I denna översyn kommer den nomenklatur som används för beskrivningen av CFTR-genmutationer som utvecklats av Human genom Variation Society15 att användas.
mutationer och Proteindefekt
CF är en autosomal recessiv ärftlig sjukdom, så mutationen måste vara närvarande i båda kopiorna av CFTR-genen som ska påverkas. Hittills har över 1900 CFTR-genmutationer associerade med sjukdomen identifierats i kodningssekvensen, messenger RNA eller andra element. Mutationer i CFTR-genen är tillgängliga för samråd i Cystic Fibrosis Mutation Database.,16 den första mutationen som beskrivs, och den vanligaste världen över, är Phe508del, men det finns andra specifika mutationer med varierande frekvens bland olika etniska grupper. I Spanien, den genomsnittliga frekvensen av Phe508del mutation är mellan 50% och 60% av alla studerade kromosomer, den näst vanligaste är Gly542X med 4%-8%, följt av Asn1303Lys i 2%-4% av fallen. De mutationer som hittills beskrivits klassificeras i sex typer eller klasser enligt den mekanism som orsakar sjukdomen.17 dessa typer av mutationer sammanfattas i Fig. 1., Klass i-mutationer leder till en för tidig stoppkodon i messenger RNA som förhindrar översättning av det fullständiga proteinet. Således är det producerade proteinet kort och icke-fungerande. Klass II-mutationer kodar för ett strukturellt onormalt och misfoldat protein som avlägsnas av endoplasmatisk retikulum innan det når cellytan. Den vanligaste mutationen i CF, Phe508del, tillhör denna grupp. Vid mutationer i klasserna III till VI når proteinerna cellytan, men fungerar inte ordentligt. Mutationer av klass III orsakar minskad kanalaktivering, så kanalerna förblir stängda., Klass IV-mutationer orsakar en minskning av jonledningsförmågan genom kanalen. Klass V-mutationer kodar för mindre proteiner som resulterar i en reducerad mängd CFTR i cellytan, så att en viss funktion uppträder, men på en reducerad nivå. Slutligen leder klass VI-mutationer till en förkortad halveringstid på grund av proteininstabilitet och kan också skada regleringen av närliggande CFTR-kanaler i cellytan.
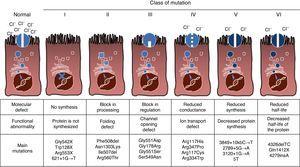
typer av mutationer vid cystisk fibros.
CFTR-modulatorer
tre huvudklasser har identifierats i utvecklingen av läkemedel för att reparera CFTR-protein.11 den första gruppen är för tidiga stopp codon suppressors (klass i mutationer). Dessa läkemedel förhindrar identifiering av denna för tidiga stoppkodon, så att proteinsyntesen kan fortsätta tills den är klar. Den andra gruppen är CFTR-korrektorerna. Dessa föreningar är utformade för att korrigera defekter vid transport av vikta protein (klass II-mutationer) till cellmembranet, där det kan fungera nästan normalt., Den tredje gruppen består av de så kallade CFTR potentiators. Dessa är läkemedel som är utformade för att rikta in CFTR-proteinet på cellytan för att förbättra dess funktion. Således kan dessa potentiatorer agera på klass III, IV, V och VI-mutationer. För närvarande undersöks många molekyler som använder dessa olika mekanismer, varav en redan har nått marknaden: Ivacaftor (VX-770) är en CFTR-potentiator som godkändes i USA i januari 2012 för behandling av CF-patienter äldre än 6 år som har Gly551Asp-mutationen.,
Mutationsbehandlingar av klass i
cirka 10% av CF-fallen orsakas av mutationer av klass i. De första drogerna som användes för denna klass var aminoglykosider. För flera år sedan rapporterades gentamicin att ha förmågan att maskera det för tidiga stoppet kodon som förhindrar syntes av CFTR-proteinet. Detta uppnås genom införande av en aminosyra som gör det möjligt för ribosomen att fortsätta läsa genen och producera ett protein i full längd. Prekliniska studier visade att proteinet kunde syntetiseras i en musmodell och 35% av proteinfunktionen återfanns in vitro.,18,19 effekten av intravenös administrering av gentamicin utvärderades i två studier av CF-patienter med olika typer av klass i-mutationer. En genomfördes i USA hos fem patienter,20 och en annan inkluderade 18 patienter i Frankrike.21 Även om svaret var positivt varierade resultaten dock i stor utsträckning, så fördelen var inte universell. Dessutom bidrog toxicitetsproblem med aminoglykosider till en ogynnsam profil.
syntetiska alternativ är ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., Detta är en molekyl utformad för att göra det möjligt för ribosomer att läsa den genetiska informationen medan ”hoppa” den för tidiga stoppkodon, som producerar ett funktionellt CFTR-protein.Farmakokinetiken för ataluren har visats i djurmodeller och i fas II-studier. I två små preliminära studier visade 23, 24 en grupp CF-patienter som behandlades med oral ataluren förbättring av sjukdomens elektrofysiologiska abnormiteter, med en ökning av antalet celler i näsan som uttrycker proteinet på deras yta., Därefter utvärderade en annan liten studie av 19 patienter olika doser ataluren administrerat oralt var 8: e timme, med förbättringar av CFTR-aktivitet och kliniska parametrar och en bra säkerhetsprofil.25 resultaten från en klinisk fas III-studie av ataluren som ännu inte formellt hade publicerats rapporterades under 2012 års nordamerikanska konferens för cystisk fibros. Totalt 238 patienter äldre än 6 år randomiserades till ataluren 10, 10, 20mg / kg eller placebo var 8: e timme i 48 veckor., Det fanns inga signifikanta skillnader i FEV1 hos ataluren jämfört med placebo efter 48 veckors behandling (- 2, 5% ataluren vs-5, 5% placebo, p=ns). När stratifierad av kronisk nebuliserad antibiotikaanvändning var det en 6,7% skillnad i medelförändring efter 48 veckor till förmån för ataluren för patienter som inte var på tobramycin, medan det inte fanns någon skillnad i förändring i FEV1 hos dem som fick nebuliserad tobramycin. Något liknande inträffade med andelen exacerbationer, där skillnaden mellan båda grupperna var signifikant., Vid stratifiering visade patienterna i atalurengruppen som inte var på nebuliserad tobramycin en procentuell minskning av exacerbationer med 43% jämfört med placebogruppen. Nasal potentialskillnad och svettkloridtest visade ingen skillnad mellan grupper, oberoende av användningen av nebuliserade antibiotika. Författarna drog slutsatsen att fördelarna var större hos patienter som inte fick kronisk antibiotikabehandling med nebuliserad aminoglykosid, spekulerar att tobramycin och ataluren interagerade på en ribosomal nivå, producerar antagonism när de används samtidigt.,26
Mutationsbehandlingar av klass II
mutationer av klass II, som är den vanligaste mutationen av sjukdomen (Phe508del), förekommer hos ett stort antal CF-patienter, vilket gör dem till ett primärt mål i CF-forskning. Olika molekyler har studerats, varav de flesta har utvecklats av Vertex Pharmaceuticals Inc. Den första korrektorföreningen, lumacaftor (VX-809), visade god in vitro-effekt, vilket förbättrade kloridtransporten i 14%.,27 resultaten var dock något nedslående hos patienter, eftersom förbättringarna i kloridkoncentrationen i svett var mycket små (7mmol/l) och det fanns inga förändringar i nasal potential.28
effekterna av CFTR-potentiatorn ivacaftor (VX-770), ett läkemedel för klass III-mutationer (se beskrivning nedan), har också undersökts hos patienter som var homozygot för Phe508del. Resultaten från DISCOVER-studien visar att ivacaftor inte är förknippat med någon signifikant förbättring av FEV1, livskvalitet eller antalet exacerbationer jämfört med placebo.,29
eftersom lumacaftor kan hjälpa till med leveransen av Phe508del-CFTR till cellytan och ivacaftor ökar öppningstiden och kloridledningen genom epitelcellen kan förbättring av den underliggande phe508del-defekten vara möjlig med kombinationen av båda molekylerna. In vitro-studier av kombinerad ivacaftor och lumacaftor i respiratoriskt epitel med Phe508del mutation har visat att lumacaftor ökar CFTR-klorid transporter med 15% på sin egen, och när ivacaftor har lagts till transporter ökar till nästan 30%., Denna kombination av läkemedel har undersökts i en fas II-studie hos patienter med Phe508delmutation. Fullständiga resultat har ännu inte publicerats, men initiala data tyder på en positiv effekt på lungfunktionen i homozygot Fe508del, men inte i heterozygositet.30 Två kliniska prövningar hos patienter som är 12 år eller äldre homozygot för mutationen Phe508del utvecklas för att utvärdera kombination ivacaftor och lumacaftor; dessa är TRAFIKEN (NCT01807923)31 och TRANSPORT (NCT01807949)32 studier. Resultaten kan tillåta ungefär hälften av CF-befolkningen att få CFTR-modulerande terapi., Studier behövs emellertid för att utvärdera effekten av kombinationsbehandling hos phe508del heterozygota individer.
ett annat alternativ är korrigeringsföreningen VX-661, och studier pågår för närvarande. Dess effektivitet testas ensamt och i kombination med ivacaftor (VX-770) och resultaten kommer snart att finnas tillgängliga (NCT01531673).33
Klass III Mutation Behandlingar
Ivacaftor (VX-770), som är en CFTR potentiator som modulerar funktion av onormala proteiner.,Denna molekyl var ursprungligen utformad för att förbättra CFTR-funktionen i kulturer av respiratoriska epitelceller som bär en enda Gly551Asp-mutation.34 Det är det första läkemedlet som godkänts i USA och Europa för behandling av CF-patienter som bär Gly551Asp mutation. Efter att ivacaftors förmåga att förbättra kloridtransporten genom cellmembranet visades in vitro, genomfördes de första fas II-studierna med 39 patienter.35 i denna studie förbättrades CFTR-proteinfunktionen tre dagar efter påbörjad behandling och nådde kloridkoncentrationer i svett på upp till normala nivåer., Med dessa resultat följde ytterligare två kliniska studier: STRIVE-studie, hos 144 patienter i åldern 12 år eller mer36 och ENVISION-studie med 52 barn mellan 6 och 11 år.37 Båda studierna inkluderade patienter med minst en Gly551Asp mutation och FEV1 mellan 40% och 105% som ursprungligen följt under en period av 14 dagar och sedan randomiserades till att ta emot 150 mg oral ivacaftor eller placebo två gånger per dag under en period på 48 veckor., Efter 48 veckors behandling fick patienterna möjlighet att fortsätta i en longitudinell öppen studie,the PERSIST study (nct01117012), 38 i 96 veckor.
I STRIVE hade patienter i ivacaftor-gruppen en förbättring med 10, 6% i FEV1 (primär endpoint) från och med dag 15 av behandlingen, som bibehölls under 48-veckorstudien. Vidare observerades en minskning av kloridkoncentrationen i svett (medelvärde: -48,7 mmol/l), med förbättrad livskvalitet, en 55% minskning av exacerbationer och en viktökning på 2,7 kg.,
resultaten av ENVISION-studien sammanfaller i stor utsträckning med resultaten från studien hos ungdomar och vuxna, med skillnaden att livskvaliteten inte nådde statistisk skillnad. De biverkningar som mer frekvent observerades i behandlingsgruppen i både STRIVE och ENVISION studier var övre luftvägsinfektioner, nästäppa, halsont, yrsel och hudutslag., De preliminära resultaten efter de första 12 veckorna av den kvarstående studien visar att förbättringar av lungfunktionen (FEV1), andningssymtom och viktökning hos patienter som behandlas med ivacaftor bibehålls under denna period. Dessutom upplevde subgruppen av patienter som bytte från placebo till ivacaftor vid baslinjen till PERSIST en 10, 8% förbättring av FEV1 vid 15 dagar och 13% vid 12 veckor, tillsammans med en minskning av exacerbationer.,39
trots de goda resultaten med ivacaftor vid behandling av Gly551Asp-mutationen hos barn äldre än 6 år och vuxna vid 48 veckor återstår vissa problem att lösa. För det första har läkemedlet inte testats hos barn under 6 år. Det verkar dock förnuftigt att korrigera defekten innan irreversibel skada uppstår, med tanke på att lungpåverkan börjar före sex års ålder. En rättegång i denna åldersgrupp pågår för närvarande (NCT01705145).40 för det andra skulle ett annat alternativ vara att även prova andra mutationer av klass III., I detta avseende har in vitro-studier på nio andra mutationer visat mycket liknande resultat, 41 så det är rimligt att förvänta sig liknande resultat hos patienter. Det är en pågående fas III klinisk prövning på patienter som är äldre än 6 år med andra klass III mutationer (KONTINUE och KONNECTION studier. NCT01614470).42,43 för det tredje, även om det inte finns någon behandling för resten av mutationsklasserna (IV–VI), kan CFTR-potentiatorer vara lika fördelaktiga i dessa fall., En klinisk prövning genomförs i detta avseende, som utvärderar effekten av ivacaftor i Arg117His klass IV-mutation (KONDUCT studie. NCT01614457).44 slutligen har effektiviteten och den långsiktiga säkerheten för ivacaftor efter 48 veckor ännu inte fastställts. G551D-observationsstudien (GOAL; NCT01521338)45 är en observationsstudie efter patienter äldre än 6 år som fick ivacaftor., Det syftar till att rapportera effekten och säkerheten hos långsiktiga ivacaftor, tillsammans med andra resultat av intresse som inkluderar inflammatoriska mediatorer i sputum, mukociliär clearance och gastrointestinalt pH. resultaten förväntas i slutet av 2013.
slutsatser
CF är ett exempel på en sjukdom som är väl positionerad för att dra nytta av personlig medicin. För närvarande syftar två mycket olika tillvägagångssätt till att korrigera den grundläggande defekten: genterapi som syftar till att korrigera den genetiska förändringen och terapi med molekyler som syftar till att korrigera den funktionella defekten på proteinnivån., Den senare börjar visa lovande resultat för olika molekyler i utveckling, och en av dem (ivacaftor) marknadsförs redan för Gly551Asp klass III-mutation, med utmärkt resultat hos barn äldre än 6 år, ungdomar och vuxna. Det slutliga målet är att tillhandahålla korrigerings-och potentiatorbehandlingar för alla CF-patienter oavsett deras mutation. Eftersom resultaten av dessa och andra nya molekyler uppträder är det troligt att en specifik molekyl eller kombination kommer att behövas för varje patient, beroende på deras befintliga mutationer., I vilket fall som helst är framtiden lovande, och viktiga steg har tagits mot att få en behandling som effektivt kommer att agera på orsaken till denna sjukdom.
intressekonflikter
författarna förklarar inga intressekonflikter.