Introducere
finalizarea proiectului genomului uman a fost relevant piatră de hotar pentru cunoștințe medicale, oferind informațiile necesare pentru a înțelege caracteristicile unice ale fiecărui individ.1 consecința logică a acestor cunoștințe ar fi aceea de a putea aplica teste de diagnostic și tratamente specifice fiecărui pacient pe baza informațiilor genetice individuale. Această nouă formă de îngrijire medicală se numește medicină personalizată.,2 cu toate acestea, în ciuda progreselor majore care au dus la cunoașterea genomului uman, traducerea acestuia în tratamente de diagnostic și personalizate a fost mai mică decât se aștepta. În prezent, se fac pași în această direcție cu două inițiative majore: sisteme de biologie3 și farmacogenetică.,4 scopul final al acestor inițiative este de a dezvolta o practică medicală personalizată în funcție de caracteristicile fiecărui individ, care poate prezice debutul sau un curs de o anumită boală, permite adecvate strategii de prevenire să fie stabilite și, în cele din urmă, permite pacientului să ia parte la luarea deciziilor. Acesta a fost numit medicament P4.5
fibroza chistică (CF) rămâne cea mai frecventă și letală boală genetică în rândul caucazienilor., Acesta se produce la o rată de 1 la 2500-6000 nou-născuți, în funcție de regiune și origine etnică, și într-o proporție de purtători sănătoși, variind între 1:20 și 37.6 În Spania, datorită introducerea progresivă a programelor de screening neonatal în diferite comunități, o incidență mai mică a CF este acum de a fi văzut decât s-a estimat anterior, în 2009, de exemplu, 1/4430 născuți vii în Galicia, 1/4339 în Castilia-Leon, 1/5376 în Murcia, 1/5840 în Catalonia, și 1/6602 în Insulele Baleare.7 Se estimează că există 70000 de bolnavi de CF în întreaga lume.,8 boala este cauzată de mutații ale genei care codifică proteina reglatoare fibroza chistică regulator de conductanță transmembranară (CFTR), un canal de clorură implicat în eliberarea adenozin trifosfatului și reglarea altor canale de transport de ioni. Această proteină este exprimată în celulele epiteliale respiratorii, pancreas, tractul biliar, glandele sudoripare și sistemul genito-urinar., Modificarea sa duce la o anomalie în transportul ionilor, astfel încât pacienții produc mucus gros, lipicios, care înfundă canalele organului în care se află și astfel modificarea prezintă efecte multisistemice care determină gama largă de manifestări clinice ale FC. În ciuda progreselor majore în tratamentul FC care au dus la o supraviețuire mai lungă (mediana actuală este estimată la 37,5 ani) 9,există încă un drum lung de parcurs pentru a se asigura că pacienții cu FC au o cantitate și o calitate a vieții similare cu cea a subiecților fără boală., În acest context, sunt necesare noi tratamente pentru scăderea morbidității și creșterea supraviețuirii.CF este un exemplu de boală bine poziționată pentru a profita de medicina personalizată. Pe de o parte, este o boală monogenică, cauzată de mutațiile unei gene specifice. Fiziopatologia entității este bine caracterizată, iar țintele terapeutice sunt clare. Mai mult, diagnosticul bolii necesită testare genetică pentru identificarea tipului de boală, astfel încât defectul genetic exact este determinat în fiecare caz.,10
în prezent, două abordări foarte diferite sunt orientate spre corectarea defectului de bază: terapia genică, care vizează corectarea modificării genetice și terapia cu molecule, care vizează corectarea defectului funcțional la nivelul proteinei. Accentul terapiei genice este introducerea copiilor genetice normale în căile respiratorii ale pacienților cu CF. Aceasta implică introducerea unui vector viral recombinant, al cărui ADN a fost extras și înlocuit cu noul ADN terapeutic. Vectorul viral servește ca vehicul pentru introducerea noului ADN în celula țintă., Până în prezent au fost utilizate diferite tipuri de virusuri, cum ar fi adenovirusul sau lentivirusul. Mai mult, au fost dezvoltate și particule non-virale, cum ar fi nanoparticule capabile să insereze ADN.11 cu toate acestea, rezultatele de până acum au fost slabe, deoarece durata de exprimare a genei introduse a fost scurtă cu ambele tipuri de vectori.12 Consorțiul pentru terapie genică din Marea Britanie dezvoltă un studiu clinic de fază II pentru a evalua eficacitatea clinică a unui vector ADN plasmidic/lipozomal optimizat.13 obiectivul de recrutare este de 130 de pacienți, iar rezultatele sunt așteptate în 2014 (NCT01621867).,14
pe de altă parte, terapia îndreptată spre restabilirea funcției proteinei CFTR a avut mai mult succes. În ultimii ani, rezultatele încep să apară pe medicamente care acționează direct asupra proteinei CFTR. De fapt, în ianuarie 2012, primul medicament pentru corectarea defectelor de mutație Gly551Asp a fost comercializat în SUA. În secțiunile următoare vom examina informațiile disponibile privind progresul medicinii personalizate pentru FC și tratamentele disponibile pentru corectarea defectului care provoacă boala la nivelul proteinei., În această revizuire, va fi utilizată nomenclatura utilizată pentru descrierea mutațiilor genei CFTR dezvoltate de Societatea de variație a genomului Uman15.
mutații și defecte proteice
CF este o boală ereditară recesivă autosomală, astfel încât mutația trebuie să fie prezentă în ambele copii ale genei CFTR pentru a fi afectată. Până în prezent, peste 1900 de mutații ale genei CFTR asociate bolii au fost identificate în secvența de codificare, ARN mesager sau alte elemente. Mutațiile genei CFTR sunt disponibile pentru consultare în baza de date a mutațiilor fibrozei chistice.,16 prima mutație descrisă și cea mai frecventă la nivel mondial este Phe508del, dar există și alte mutații specifice cu frecvență variabilă între diferite grupuri etnice. În Spania, frecvența medie de Phe508del mutație este între 50% și 60% din totalul studiat de cromozomi, a doua cea mai frecventă este Gly542X cu 4%-8%, urmat de Asn1303Lys în 2%-4% din cazuri. Mutațiile descrise până în prezent sunt clasificate în șase tipuri sau clase în funcție de mecanismul care provoacă boala.17 aceste tipuri de mutații sunt rezumate în Fig. 1., Mutațiile de clasa I conduc la un codon Stop prematur în ARN-ul mesager care împiedică traducerea proteinei complete. Astfel, proteina produsă este scurtă și nefuncțională. Mutațiile din clasa II codifică o proteină anormală din punct de vedere structural și rătăcită, care este îndepărtată de reticulul endoplasmatic înainte de a ajunge la suprafața celulei. Cea mai frecventă mutație în CF, Phe508del, aparține acestui grup. În cazul mutațiilor din clasele III-VI, proteinele ajung la suprafața celulei, dar nu funcționează corect. Mutațiile din clasa III determină o activare scăzută a canalului, astfel încât canalele rămân închise., Mutațiile din clasa IV determină o scădere a conductanței ionice prin canal. Mutațiile din clasa V codifică proteine minore rezultând o cantitate redusă de CFTR în suprafața celulei, astfel încât apare o anumită funcție, dar la un nivel redus. În cele din urmă, mutațiile din clasa VI duc la un timp de înjumătățire scurtat din cauza instabilității proteinelor și pot deteriora, de asemenea, reglarea canalelor CFTR vecine pe suprafața celulei.
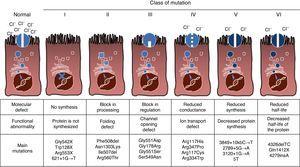
tipuri de mutații în fibroza chistică.
CFTR Modulatori
Trei clase principale au fost identificate în dezvoltarea de medicamente pentru repararea proteine CFTR.11 primul grup sunt supresoare de codon Stop prematur (mutații de clasa I). Aceste medicamente împiedică identificarea acestui codon de oprire prematură, astfel încât sinteza proteinelor să poată continua până la finalizare. Al doilea grup sunt corectorii CFTR. Acești compuși sunt concepuți pentru a corecta defectele în transportul proteinei pliate (mutații de clasa II) către membrana celulară, unde poate funcționa aproape normal., Al treilea grup constă din așa-numitele potențiatoare CFTR. Acestea sunt medicamente concepute pentru a viza proteina CFTR pe suprafața celulei pentru a-și îmbunătăți funcția. Astfel, acești potențiatori pot acționa asupra mutațiilor din clasa III, IV, V și VI. În prezent, numeroase molecule folosind aceste mecanisme diferite sunt în curs de investigare, dintre care unul a ajuns deja pe piață: Ivacaftor (VX-770) este un CFTR potențator aprobat în SUA în ianuarie 2012 pentru tratamentul CF pacienții cu vârsta peste 6 ani care au Gly551Asp mutație.,
tratamente de mutație clasa I
aproximativ 10% din cazurile de FC sunt cauzate de mutații de clasa I. Primele medicamente utilizate pentru această clasă au fost aminoglicozidele. Cu câțiva ani în urmă, s-a raportat că gentamicina are capacitatea de a masca codonul de oprire prematură care împiedică sinteza proteinei CFTR. Acest lucru se realizează prin introducerea unui aminoacid care permite ribozomului să continue citirea genei, producând o proteină de lungime întreagă. Studiile preclinice au demonstrat că proteina ar putea fi sintetizată într-un model de șoarece și 35% din funcția proteică a fost recuperată in vitro.,18,19 efectul administrării intravenoase de gentamicină a fost evaluat în două studii la pacienți cu FC cu diferite tipuri de mutații de clasa I. Unul a fost efectuat în SUA la cinci pacienți,20 și altul a inclus 18 pacienți în Franța.21 Cu toate acestea, deși răspunsul a fost pozitiv, rezultatele au variat foarte mult, astfel încât beneficiul nu a fost universal. Mai mult, problemele de toxicitate cu aminoglicozidele au contribuit la un profil nefavorabil.o alternativă sintetică este ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, SUA)., Aceasta este o moleculă concepută pentru a permite ribozomilor să citească informațiile genetice în timp ce „sărind” codonul Stop prematur, producând o proteină CFTR funcțională.Farmacocinetica atalurenului a fost demonstrată pe modele animale și în studii de fază II. În două mici studii preliminare,23,24, un grup de CF pacienții tratați pe cale orală cu ataluren au prezentat o ameliorare de anomalii electrofiziologice ale bolii, cu o creștere a numărului de celule în nas exprimarea proteinelor de pe suprafața lor., Ulterior, un alt studiu mic de 19 pacienți evaluați diferite doze de ataluren administrat pe cale orală la fiecare 8h, cu îmbunătățiri în CFTR activitate și parametrii clinici și un profil de siguranță bun.25 rezultatele unui studiu clinic de fază III cu ataluren care nu a fost încă publicat oficial au fost raportate în cadrul conferinței de fibroză chistică din America de Nord din 2012. Un total de 238 de pacienți cu vârsta peste 6 ani au fost randomizați pentru a primi ataluren 10, 10, 20 mg/kg sau placebo la fiecare 8h timp de 48 de săptămâni., Nu au existat diferențe semnificative în FEV1 în ataluren comparativ cu placebo după 48 de săptămâni de tratament (-2.5% ataluren vs -5.5% pentru placebo, P=ns). Când stratificat de cronică nebulizată utilizarea de antibiotice, nu a fost de 6,7%, diferența în modificarea medie după 48 de săptămâni în favoarea ataluren pentru pacienții care nu tobramicina, în timp ce nu există nici o diferență în modificarea FEV1 la cei care primesc nebulizată tobramicina. Ceva similar a avut loc cu procentul de exacerbări, unde diferența dintre ambele grupuri a fost semnificativă., Când stratificat, pacienții în ataluren grup nu pe nebulizată tobramicina a arătat o scădere procentuală în exacerbari de 43%, comparativ cu grupul placebo. Diferența de potențial nazal și testele de clorură de transpirație nu au arătat nicio diferență între grupuri, indiferent de utilizarea antibioticelor nebulizate. Autorii au concluzionat că beneficiile au fost mai mari la pacienții care nu au primit cronice terapia cu antibiotice cu nebulizată aminoglicozide, speculând că tobramicina și ataluren interacționat la un nivel ribozomal, care produc antagonism atunci când sunt utilizate simultan.,26
tratamente de mutație clasa II
mutațiile de clasa II, fiind cea mai frecventă mutație a bolii (Phe508del), sunt prezente la un număr mare de pacienți cu FC, făcându-i o țintă principală în cercetarea FC. Au fost studiate diverse molecule, dintre care majoritatea au fost dezvoltate de Vertex Pharmaceuticals Inc. Primul compus corector, lumacaftor (VX-809), a arătat o bună eficacitate in vitro, îmbunătățind transportul clorurilor în 14%.,27 Cu toate acestea, rezultatele au fost oarecum dezamăgitoare la pacienți, deoarece îmbunătățirile concentrației de clorură în transpirație au fost foarte mici (7mmol/l) și nu au existat modificări ale potențialului nazal.28
efecte ale CFTR potențator ivacaftor (VX-770), un medicament pentru clasa a III-mutații (a se vedea descrierea de mai jos), de asemenea, au fost investigate la pacienții care au fost homozigote pentru Phe508del. Rezultatele studiului DISCOVER arată că ivacaftor nu este asociat cu o îmbunătățire semnificativă a FEV1, a calității vieții sau a numărului de exacerbări comparativ cu placebo.,29
Începând cu lumacaftor poate ajuta livrarea de Phe508del-CFTR la suprafața celulei și ivacaftor crește timpul de deschidere și de clorură de conducție prin celulelor epiteliale, îmbunătățirea de fond al sistemului Phe508del defect poate fi posibil cu o combinație de ambele molecule. Studiile In vitro de combinat ivacaftor și lumacaftor în epiteliul respirator cu Phe508del mutație au arătat că lumacaftor crește CFTR clorură de transport cu 15% pe cont propriu, și ivacaftor atunci când este adăugat, transport crește la aproape 30%., Această combinație de medicamente a fost investigată într-un studiu de fază II la pacienții cu mutație Phe508del. Rezultatele complete nu au fost încă publicate, dar datele inițiale sugerează un efect benefic asupra funcției pulmonare în Phe508del homozigot, dar nu și în heterozigozitate.30 Două studii clinice la pacienții 12 ani sau mai în vârstă homozigot pentru mutația Phe508del sunt dezvoltate pentru a evalua combinate ivacaftor și lumacaftor; acestea sunt de TRAFIC (NCT01807923)31 și TRANSPORT (NCT01807949)32 studii. Rezultatele ar putea permite aproximativ jumătate din populația FC să primească terapie modulatoare CFTR., Cu toate acestea, sunt necesare studii pentru a evalua efectul terapiei combinate la indivizii heterozigoți Phe508del.o altă alternativă este compusul corector VX-661, iar studiile sunt în curs de desfășurare. Eficacitatea sa este testată singură și în asociere cu ivacaftor (VX-770), iar rezultatele vor fi disponibile în curând (NCT01531673).Ivacaftor (VX-770) este un potențiator CFTR care modulează funcția proteinei anormale.,34 această moleculă a fost inițial concepută pentru a îmbunătăți funcția CFTR în culturile celulelor epiteliale respiratorii care poartă o singură mutație Gly551Asp.34 este primul medicament aprobat în SUA și Europa pentru tratamentul CF la pacienții care poartă mutația Gly551Asp. După ce capacitatea ivacaftor de a îmbunătăți transportul clorurii prin membrana celulară a fost demonstrată in vitro, primele studii de fază II au fost efectuate la 39 de pacienți.35 în acest studiu, funcția proteinei CFTR s-a îmbunătățit la trei zile după începerea tratamentului, atingând concentrații de clorură în transpirație până la valori normale., Cu aceste rezultate, au urmat două studii clinice suplimentare: studiul STRIVE, la 144 de pacienți cu vârsta de 12 ani sau mai mult36 și studiul ENVISION, care a inclus 52 de copii cu vârsta cuprinsă între 6 și 11 ani.37 Ambele studii au inclus pacienți cu cel puțin una Gly551Asp mutație și FEV1 între 40% și 105%, care inițial au fost urmate de o perioadă de 14 zile și apoi randomizați să primească 150 mg oral ivacaftor sau placebo de două ori pe zi pentru o perioadă de 48 de săptămâni., După încheierea celor 48 de săptămâni de tratament, pacienților li s-a oferit posibilitatea de a continua într-un studiu longitudinal deschis, studiul PERSIST (NCT01117012),38 timp de 96 de săptămâni.
În STRADUIM, pacienții în ivacaftor grup a avut o îmbunătățire de 10,6% FEV1 (obiectiv final principal) incepand din a 15-a zi de tratament, care s-a menținut timp de 48 săptămâni de studiu. Mai mult, a fost observată o scădere a concentrației de clorură în transpirație (medie: -48,7 mmol/l), cu o calitate îmbunătățită a vieții, o reducere cu 55% a exacerbărilor și o creștere în greutate de 2,7 kg.,rezultatele studiului ENVISION coincid în mare măsură cu cele ale studiului la adolescenți și adulți, cu diferența că calitatea vieții nu a atins diferența statistică. Efectele adverse observate mai frecvent în grupul de tratament, atât în studiile STRIVE, cât și în cele ENVISION, au fost infecții ale tractului respirator superior, congestie nazală, dureri în gât, amețeli și erupții cutanate., Rezultatele preliminare după primele 12 săptămâni ale studiului PERSIST arată că ameliorarea funcției pulmonare (FEV1), simptomele respiratorii și creșterea în greutate în rândul pacienților tratați cu ivacaftor se mențin în această perioadă. Mai mult decât atât, la subgrupul de pacienți care au trecut de la placebo la ivacaftor la bază PERSISTĂ în a cunoscut o 10.8% ameliorarea FEV1 la 15 zile, și 13% la 12 săptămâni, împreună cu o reducere în exacerbări.,39
în Ciuda rezultatelor bune cu ivacaftor în tratamentul Gly551Asp mutație la copii cu vârsta peste 6 ani și adulți la 48 de săptămâni, unele probleme rămân a fi rezolvate. În primul rând, medicamentul nu a fost testat la copii sub 6 ani. Cu toate acestea, pare sensibil să corectăm defectul înainte de apariția leziunilor ireversibile, având în vedere că implicarea pulmonară începe înainte de vârsta de șase ani. Un proces în acest interval de vârstă este în curs de desfășurare (NCT01705145).40 În al doilea rând, o altă alternativă ar fi încercarea altor mutații din clasa III., În acest sens, studiile in vitro pe alte nouă mutații au arătat rezultate foarte similare, 41 Deci este rezonabil să se aștepte rezultate similare la pacienți. Există un studiu clinic de fază III în desfășurare la pacienți cu vârsta peste 6 ani cu alte mutații de clasa III (studii KONTINUE și KONNECTION; NCT01614470).42,43 în al treilea Rând, deși nici un tratament nu este disponibil pentru restul de mutație clase (IV–VI), CFTR potențiatori poate fi la fel de benefice în aceste cazuri., Un studiu clinic este efectuat în acest sens, evaluarea eficacității ivacaftor în Arg117His clasa a IV-mutația (KONDUCT studiu; NCT01614457).În cele din urmă, eficacitatea și siguranța pe termen lung a ivacaftor după 48 de săptămâni nu au fost încă stabilite. La G551D Studiu Observațional (OBIECTIV; NCT01521338)45 este un studiu observațional următoarele pacienții cu vârsta peste 6 ani primesc ivacaftor., Acesta vizează raportarea eficacității și siguranței ivacaftor pe termen lung, împreună cu alte rezultate de interes care includ mediatori inflamatori în spută, clearance-ul mucociliar și pH-ul gastrointestinal. rezultatele sunt așteptate la sfârșitul anului 2013.
concluzii
CF este un exemplu de boală bine poziționată pentru a profita de medicina personalizată. În prezent, două abordări foarte diferite vizează corectarea defectului de bază: terapia genică care vizează corectarea modificării genetice și terapia cu molecule care vizează corectarea defectului funcțional la nivelul proteinei., Acesta din urmă a început să arate rezultate promițătoare pentru diverse molecule în dezvoltare, iar una dintre ele (ivacaftor) este deja comercializat pentru Gly551Asp clasa III mutație, cu rezultate excelente la copii cu vârsta peste 6 ani, adolescenți și adulți. Obiectivul final este de a oferi tratamente corectoare și potențiatoare pentru toți pacienții cu FC, indiferent de mutația lor. Pe măsură ce apar rezultatele acestor și ale altor molecule noi, este probabil ca o moleculă sau o combinație specifică să fie necesară pentru fiecare pacient, în funcție de mutațiile existente., În orice caz, viitorul este promițător și s-au luat măsuri importante pentru obținerea unui tratament care să acționeze eficient asupra cauzei acestei boli.
conflicte de interese
autorii nu declară conflicte de interese.