ile genów znajduje się w genomie?
Tryb Reader
zbadaliśmy już wielką różnorodność rozmiarów genomów w całym świecie żywym (patrz tabela w winiecie NA „How big are genomes?”). Jako pierwszy krok w udoskonalaniu naszego zrozumienia zawartości informacji w tych genomach, potrzebujemy poczucia liczby genów, które przechowują. Kiedy mówimy o genach, będziemy myśleć o genach kodujących białka, wykluczając wciąż rozszerzający się zbiór regionów kodujących RNA w genomach.,
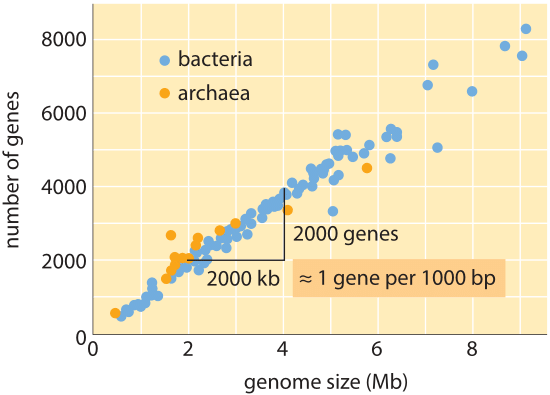
Rysunek 1: liczba genów jako funkcja wielkości genomu. Rysunek pokazuje dane dla różnych bakterii i archeea, z nachyleniem linii danych potwierdzających prostą regułę kciuka dotyczące wielkości genomu i liczby genów. (Adaptacja z M. Lynch, początki architektury genomu.,)
na całym drzewie życia, chociaż rozmiary genomu różnią się aż o 8 rzędów wielkości (od<2 kb dla wirusa zapalenia wątroby typu D (BNID 105570) do>100 Gbp dla Marmurkowca (BNID 100597) i niektórych kwiatów Fritillaria (BNID 102726)), zakres liczby genów waha się o mniej niż 5 rzędów wielkości (od wirusów takich jak MS2 i QB bakteriofagi posiadające tylko 4 geny do około stu tysięcy w pszenicy). Wiele bakterii ma kilka tysięcy genów., Zawartość tego genu jest proporcjonalna do wielkości genomu i wielkości białka, jak pokazano poniżej. Co ciekawe, genomy eukariotyczne, które są często tysiąc razy lub więcej większe niż u prokariotów, zawierają tylko rząd wielkości więcej genów niż ich prokariotyczne odpowiedniki. Niemożność skutecznego oszacowania liczby genów u eukariotów na podstawie wiedzy o zawartości genów prokariotów była jednym z nieoczekiwanych zwrotów akcji współczesnej biologii.,
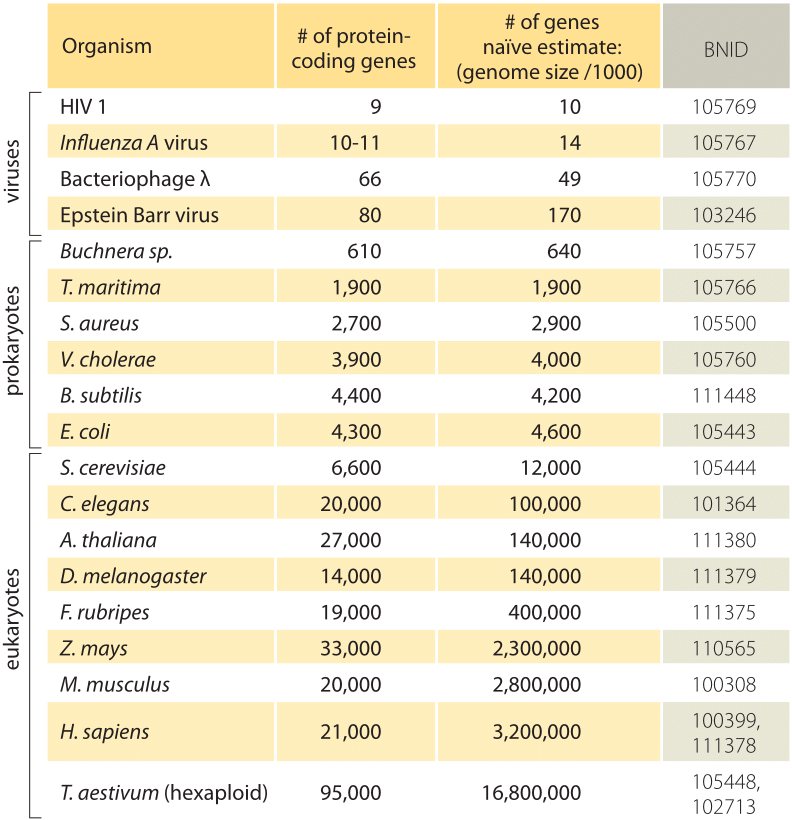
Table 1: A comparison between the number of genes in a organism and a naiwny estymate based on the genome size divided by a constant factor of 1000bp/Gen, tj. predicted number of genes = Genome size / 1000. Okazuje się, że ta surowa zasada działa zaskakująco dobrze dla wielu bakterii i archeea, ale nie nędznie dla organizmów wielokomórkowych.
najprostsze oszacowanie liczby genów w genomie opiera się na założeniu, że całość genomu koduje geny będące przedmiotem zainteresowania., Aby poczynić dalsze postępy z szacunkami, musimy mieć miarę liczby aminokwasów w typowym białku, które przyjmiemy do około 300, wiedząc jednak, że podobnie jak genomy, białka występują w różnych rozmiarach, jak ujawniono w winiecie na ten temat, ” jakie są rozmiary białek?”. Na podstawie tego skromnego założenia widzimy, że liczba zasad potrzebnych do kodowania naszego typowego białka wynosi około 1000 (3 pary zasad na aminokwas)., W związku z tym, w ramach tego sposobu myślenia, liczba genów zawartych w genomie jest szacowana na rozmiar genomu/1000. W przypadku genomów bakteryjnych strategia ta działa zaskakująco dobrze, co widać w tabeli 1 i Rysunku 1. Na przykład, gdy stosuje się do E. coli K-12, genomu 4,6 x 106 bp, ta zasada prowadzi do oszacowania 4600 genów, które można porównać do obecnej najlepszej wiedzy o tej ilości, która wynosi 4225. W przechodzeniu przez kilkanaście reprezentatywnych bakterii i archeal genomów w tabeli podobnie uderzające predykcyjne moc do około 10% jest obserwowana., Z drugiej strony, strategia ta zawodzi spektakularnie, gdy zastosujemy ją do genomów eukariotycznych, co skutkuje na przykład oszacowaniem, że liczba genów w ludzkim genomie powinna wynosić 3 000 000, co jest zawyżeniem. Zawodność tego oszacowania pomaga wyjaśnić istnienie puli zakładów Genesweep, która już na początku 2000 roku miała ludzi obstawiających liczbę genów w ludzkim genomie, przy szacunkach ludzi zmieniających się o więcej niż dziesięciokrotnie.,
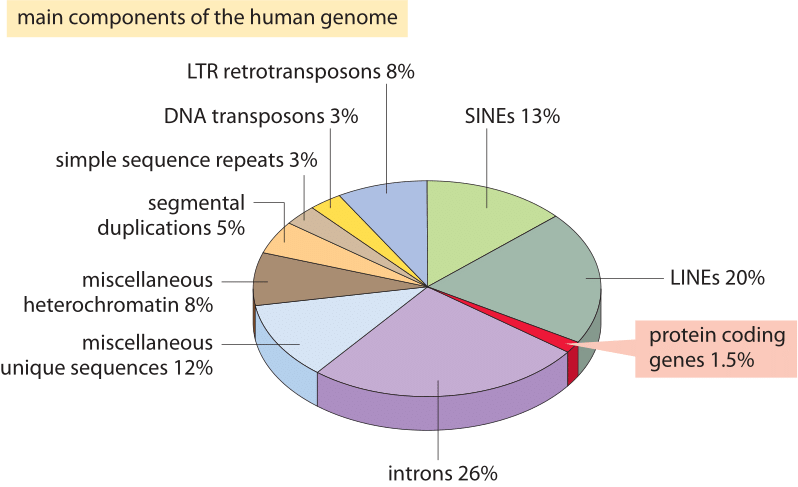
Rysunek 2: różne składniki sekwencji składające się na ludzki genom. Około 1,5% genomu składa się z ≈20 000 sekwencji kodujących białka, które są przeplatane przez niekodujące introny, co stanowi około 26%. Pierwiastki transponowalne są największą frakcją (40-50%), obejmującą np. długie przeplatane pierwiastki jądrowe (linie) i krótkie przeplatane pierwiastki jądrowe (SINEs). Większość transponowalnych elementów to pozostałości genomowe, które są obecnie nieistniejące. (BNID 110283, adaptowany z T. R. Gregory Nat Rev Genet., 9: 699-708, 2005 na podstawie International Human Genome Sequencing Consortium. Wstępne sekwencjonowanie i analiza ludzkiego genomu. Przyroda 409: 860 2001.)
co tłumaczy tę spektakularną porażkę najbardziej naiwnego oszacowania i czego nas uczy o informacji zorganizowanej w genomach? Genomy eukariotyczne, zwłaszcza te związane z organizmami wielokomórkowymi, charakteryzują się szeregiem intrygujących cech, które zaburzają prosty obraz kodujący wykorzystany w naiwnej ocenie., Te różnice w wykorzystaniu genomu są przedstawione Obrazkowo na rysunku 2, który pokazuje procent genomu używanego do innych celów niż kodowanie białka. Jak widać na rysunku 1, prokaryotes mogą efektywnie kompaktować swoje sekwencje kodujące białka tak, że są prawie ciągłe i powodują, że mniej niż 10% ich genomów jest przypisane do niekodującego DNA (12% W E. coli, BNID 105750), podczas gdy u ludzi ponad 98% (BNID 103748) jest niekodującym DNA.,
odkrycie tych innych zastosowań genomu stanowi jedne z najważniejszych wglądów w DNA, a bardziej ogólnie w biologię, z ostatnich 60 lat. Jednym z tych alternatywnych zastosowań dla nieruchomości genomicznych jest Genom regulacyjny, a mianowicie sposób, w jaki duże kawałki genomu są wykorzystywane jako cele do wiązania białek regulatorowych, które powodują kombinatoryczną kontrolę tak typową dla genomów w organizmach wielokomórkowych., Inną z kluczowych cech genomów eukariotycznych jest organizacja ich genów w introny i egzony, przy czym wyrażone egzony są znacznie mniejsze niż interweniujące i splatane introny. Poza tymi cechami istnieją endogenne retrowirusy, kopalne Relikty dawnych infekcji wirusowych i uderzająco, ponad 50% genomu jest zajęte przez istnienie powtarzających się elementów i transpozonów, których różne formy mogą być interpretowane jako samolubne geny, które mają mechanizmy rozmnażania się w genomie gospodarza., Niektóre z tych powtarzających się elementów i transpozonów są nadal aktywne do dziś, podczas gdy inne pozostały reliktem po utracie zdolności do dalszego proliferacji w genomie.
podsumowując, genomy można podzielić na dwie główne klasy: zwartą i ekspansywną. Te pierwsze są gęste genowo, z zaledwie około 10% niekodującego regionu i ścisłą proporcjonalnością między wielkością genomu a liczbą genomu. Grupa ta obejmuje genomy o wielkości do około 10 Mbp, obejmujące wirusy, bakterie, archeony i niektóre jednokomórkowe eukarioty., Ta ostatnia klasa nie wykazuje wyraźnej korelacji między wielkością genomu a liczbą genów, składa się głównie z elementów niekodujących i obejmuje wszystkie organizmy wielokomórkowe.




