wprowadzenie
czynnik jądrowy kappa B (NF-kB) jest starożytnym czynnikiem transkrypcyjnym białek (Salminen et al., 2008) i uważany za regulatora odporności wrodzonej (Baltimore, 2009). Szlak sygnałowy NF-kB łączy sygnały patogenne i sygnały zagrożenia komórkowego, organizując w ten sposób odporność komórkową na inwazyjne patogeny., W rzeczywistości wiele badań wykazało, że NF-kB jest węzłem sieciowym odpowiedzialnym za złożoną sygnalizację biologiczną (Albensi and Mattson, 2000; Kaltschmidt and Kaltschmidt, 2009; Karin, 2009). W tym celu postawiono hipotezę, że NF-kB jest głównym regulatorem ewolucyjnie zachowanych kaskad biochemicznych (Mattson et al., 2000). Inne czynniki są również translokowane do mitochondriów i biorą udział w modulacji ekspresji (Barshad et al., 2018a), ale nie są przedmiotem tej recenzji., Celem niniejszej recenzji jest próba zrozumienia, w jaki sposób aktywność NF-kB przyczynia się do funkcji mitochondriów. Zakłada się, że czytelnik ma już zrozumienie podstawowej biologii mitochondrialnej. W przypadku dalszych badań czytelnik odnosi się do wielu doskonałych badań i recenzji dotyczących struktury i funkcji mitochondriów (Hall, 1979; Fox, 1982; Roger and Silberman, 2002; Henze and Martin, 2003; Conradt, 2006; Ettema, 2016; Wang and Youle, 2016; Barshad et al., 2018b).,
Aktywacja NF-kB
podjednostki czynnika jądrowego kappa B, składające się z kompleksu NF-kB, ulegają ekspresji zarówno w neuronach, jak i glejach. Kompleks NF-kB istnieje w stanie nieaktywnym w cytoplazmie (Ghosh et al., 1998; Aggarwal et al., 2004; Hayden and Ghosh, 2004), gdzie aktywacja NF-kB została dobrze opisana (Li and Karin, 2000; Baud and Jacque, 2008; Izrael, 2010). Po pobudzeniu przez cząsteczki takie jak TNFa lub inne stresory komórkowe, TNFa wiąże się z receptorami TNF (ryc. 1)., Wiązanie to, poprzez kilka etapów pośrednich, prowadzi do interakcji z kompleksem kinazy IkB (IKK), która następnie prowadzi do fosforylacji IkB, a następnie prowadzi do ubikwitynacji i degradacji IkB. Po degradacji Pozostałe dimery NF-kB (np. podjednostki p65/p50) translokują się do jądra, gdzie wiążą się z sekwencją konsensusu DNA różnych genów docelowych. Selektywność odpowiedzi NF-kB opiera się na kilku czynnikach (Sen and Smale, 2010), w tym składzie dimeru, czasie i typie komórki., Wpływ NF-kB na przeżycie komórek jest również złożony i może być neuroprotekcyjny lub prozapalny, w zależności od typu komórki, stadium rozwoju i stanu patologicznego (Qin et al., 2007).
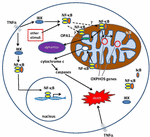
Rysunek 1. Szlaki sygnalizacji czynnika jądrowego κ B (NF-kB) w cytoplazmie i mitochondriach. Kompleks tri-subunt NF-kB (np. p65, P50, IkB – jedna z możliwych kombinacji) występuje w stanie nieaktywnym w cytoplazmie. Aktywacja NF-kB jest inicjowana, gdy cząsteczki takie jak TNFa wiążą się z receptorami TNF (istnieją różne typy)., Inne bodźce zewnętrzne lub wewnętrzne mogą również aktywować NF-kB. Po aktywacji receptorów TNF rozpoczyna się skomplikowany proces transdukcji sygnału; kinaza IkB (IKK) jest ostatecznie wyzwalana i prowadzi do fosforylacji IkB, co powoduje ubikwitynację i degradację IkB. Po degradacji IkB, możliwe jest przeniesienie pozostałych dimerów NF-kB (np. kombinacji podjednostek p65/p50 lub p50/p50) do jądra, gdzie wiąże się z sekwencją zgodną z DNA genów docelowych., Przez procesy nie dobrze poznane, kompleks NF-kB lub podjednostki NF-kB mogą również migrować do mitochondriów, gdzie dowody sugerują, że zajmują przestrzeń międzybłonową. W mitochondriach uważa się, że NF-kB wchodzi w interakcje z genami OXPHOS (mitochondrialna mtDNA), co prowadzi do ekspresji białek zaangażowanych w różne funkcje, w tym dynamikę mitochondrialną i regulację COX III (składnik kompleksu IV)., Dowody sugerują również, NF-kB może funkcjonować jako przełącznik w mitochondriach i kontrolować równowagę między wykorzystaniem glikolizy cytoplazmatycznej i oddychania mitochondrialnego w normalnych komórkach i w raku. Wreszcie, dane wskazują również na wewnętrzną stymulację szlaku apoptotycznego, gdzie aktywacja NF-kB w mitochondriach prowadzi do uwalniania cytochromu c, powodując w ten sposób kaskady kaspazy i zaprogramowaną śmierć komórki.,
organizacyjnie, NF-kB jest czynnikiem transkrypcyjnym rodziny Rel i jest związany z pięcioma genami, NF-kB1, NF-kB2, RELA, RELB i rel (Chen And Greene, 2004); geny te kodują kilka białek, NF-kB1, NF-kB2, RelA, RelB i C-rel, odpowiednio, gdzie dwa z tych białek są duże prekursorowe białka znane jako P105 i P100, które ulegają proteolizie, przekształcając się odpowiednio w P50 i P52. Białka te zawierają domeny rel-homologiczne (RHD) w swoim regionie Amino-końcowym; region RHD składa się z 2 oddzielnych, ale przylegających domen., Sekwencja najbardziej odległa od karboksy-końcowego regionu pozwala białkowi wiązać się z DNA. Bardziej wewnętrzna Sekwencja pozwala białkom rodziny rel dimeryzować (homo-lub heterodimery) w celu tłumienia ekspresji poprzez wiązanie ich odpowiedniej rodziny inhibitorów, białek IkB (Chen and Greene, 2004). Ta ostatnia Sekwencja obejmuje sekwencję lokalizacji jądrowej (NLS), która zostaje zdemaskowana, gdy IkB jest niezwiązany przez degradację. NLS ma zadanie prowadzenia lub tagowania aktywnych białek do importu do jądra komórkowego (Chen And Greene, 2004; Karin et al., 2004; Barger et al.,, 2005).
trzy z tych białek (RelA, RelB i C-REL) kodują również domenę transactivation (tad) w swoim karboksy-końcowym regionie. TADs umożliwiają tym białkom interakcję z aparatem transkrypcji podstawowej, znanym jako białko wiążące TATA (TBP), czynnik transkrypcyjny IIB, a także P300 i białko wiążące cAMP (CBP) (Chen And Greene, 2004)., Tylko te trzy białka są w stanie indukować transkrypcję swoich regionów kodujących DNA, podczas gdy inne białka, homodimery p50 i p52, są w stanie zajmować miejsca wiązania DNA bez inicjowania transkrypcji. Biorąc to pod uwagę, późniejsze 2 białka homodimerów p50 i p52 działają jako represory transkrypcyjne (Chen And Greene, 2004).
homodimery p105 i p100 zajmują miejsca wiązania DNA, blokując w ten sposób transkrypcję za pomocą czynników transkrypcyjnych, które posiadają TADs (Barger et al., 2005). Trzecią formą represji transkrypcyjnej są białka IkB., Białka te mają kilka powtórzeń ankyrin jako ich domenę rdzenia i funkcję poprzez wiązanie z RHD, które maskują NLS (Karin et al., 2004). Bez aktywnego NLS, białka NF-kB są ograniczone do cytoplazmy i nie są w stanie migrować do jądra, a więc transkrypcja jest zablokowana.
NF-kB znajduje się w mitochondriach
(2001) stwierdzono IkBa i podjednostkę NF-kB p65 we frakcjach subkomórkowych i oczyszczonych mitochondriach z komórek Jurkata. Komórki jurkata są nieśmiertelną linią komórek ludzkich limfocytów T, które są wykorzystywane do badania białaczki., W badaniu Bottero ustalono, że IkBa i NF-kB p65 zostały zlokalizowane w przestrzeni międzybłonowej mitochondrialnej. Mitochondrialna przestrzeń międzymięśniowa-przestrzeń pomiędzy wewnętrzną błoną mitochondrialną (IMM) a zewnętrzną błoną mitochondrialną (OMM).
(2003) wykazał również, że podjednostki NF-kB, p50 i p65 oraz IkBa, zostały znalezione w mitochondriach. Aby to ustalić, zastosowano kilka metod w celu dostarczenia dowodów, w tym mikroskopię elektronową odcinków komórek U937., Komórki U937 zostały po raz pierwszy wyizolowane z chłoniaka mężczyzny w średnim wieku w celu zbadania zachowania i różnicowania monocytów. Tutaj Cogswell et al. (2003) był w stanie wizualizować podjednostki NF-kB p50 i p65 oraz IkBa w wewnętrznej matrycy mitochondriów. W tym badaniu zbadano również komórki wątroby szczura oraz zidentyfikowano podjednostkę p50 i podjednostkę IkBa. Dodatkowo komórki U937 były stymulowane przez 1 h TNFa, znanym wyzwalaczem szlaku sygnałowego NF-kB., W tym eksperymencie Analiza Western blot w mitochondrialnych i cytoplazmatycznych frakcjach wykazała, że leczenie TNFa spowodowało utratę IkBa w mitochondrialnych i cytoplazmatycznych przedziałach o 30 min po leczeniu, co sugeruje, że IkBa został zdegradowany. Analizę EMSA, test in vitro wykrywający aktywację NF-kB i niespecyficzne wiązanie z sekwencjami DNA, przeprowadzono również na białku pobranym z ekstraktów jądrowych z mitochondriów izolowanych z komórek u937 stymulowanych TNFa., Stwierdzono, że sygnalizacja TNFa prowadzi do zwiększenia aktywności wiązania DNA NF-kB p50 w białku pobranym z mitochondriów.
w innych badaniach Wykryto również NF-kB w mitochondriach. Należą do nich badania (Guseva et al., 2004; Zamora et al., 2004) w liniach komórkowych ludzkiego fibroblastu HT1080, liniach komórkowych ludzkiej prostaty lncap i PC3 oraz komórkach HeLa. W komórkach LNCaP stwierdzono, że mitochondria NF-kB p50 i p65 są związane z mitochondrialnym DNA (mtDNA)., Łącznie badania te pokazują dowody na sygnalizację NF-kB w mitochondriach i że NF-kB reguluje ekspresję mitochondrialnego mRNA (patrz sekcja NF-kB i ekspresja genu mitochondrialnego poniżej).
NF-kB kontroluje dynamikę mitochondriów
istnieje kilka białek zaangażowanych w dynamikę (rozszczepienie i fuzja) i morfologię mitochondriów (Karbowski and Youle, 2003; Olichon et al., 2006; Brooks and Dong, 2007; Song et al., 2008; Autret and Martin, 2010; Silva et al., 2013; Sinha i Manoj, 2019). Jednym z nich jest atrofia optyczna 1 białka (OPA1) (Olichon et al.,, 2006; Garcia et al., 2018; Lee and Yoon, 2018). Badania sugerują, że OPA1 jest regulatorem fuzji mitochondrialnej błony wewnętrznej, a także przebudowy mitochondrialnej cristae (Cipolat et al., 2006). Ostatnio Laforge et al. (2016) wykazał, że brak IKKa miał wpływ na ekspresję OPA1 w mitochondriach i na morfologię mitochondriów.
(2017) stwierdzono, że stymulacja receptora TNFa 2 (TNFR2) Promuje fuzję mitochondrialną poprzez zależną od NF-kB aktywację ekspresji opa1 w miocytach serca., Co ważne, aktywacja TNFR2 w tym badaniu chroniła miocyty serca przed stresem poprzez zwiększenie ekspresji OPA1. Podawanie niskich stężeń egzogennego TNFa (0,5 ng/mL) przed reperfuzją niedokrwienną prowadziło do zwiększenia przeżywalności komórek, natomiast wyższe stężenia (10-20 ng / mL) prowadziło do działania toksycznego w komórkach.
NF-kB i apoptoza w mitochondriach
rola mitochondriów w programowanej śmierci komórki, czyli apoptoza jest znana od dłuższego czasu (Green and Reed, 1998; Wang and Chen, 2015)., Najważniejszą rolą mitochondriów jest wytwarzanie ATP, jednak drugą najważniejszą funkcją mitochondriów jest prawdopodobnie kontrolowanie śmierci komórek. Jak mitochondrion to robi? Jeśli mitochondrion zawodzi w wywołaniu śmierci komórki, rak jest często konsekwencją. Tak więc, w celu regulacji śmierci komórki, mitochondria integrują sygnały z różnych źródeł, które są znane jako wewnętrzne ścieżki apoptozy. Składniki aktywności NF-kB wydają się być jednym z tych sygnałów, chociaż TNFa, aktywator NF-kB, jest częścią zewnętrznego szlaku apoptozy., Szlaki zewnętrzne (za pośrednictwem receptora śmierci) są inicjowane poza komórką, podczas gdy wewnętrzne szlaki apoptozy są pośredniczone i wyzwalane w mitochondriach.
w ostatnim badaniu Pazarentzos et al. (2014) stwierdzono, że IkBa wywiera działanie pro-apoptotyczne, ponieważ hamuje anty-apoptotyczny NF-kB. W większości komórek, aktywacja NF-kB prowadzi do dalszej ekspresji genu docelowego, który wyzwala odporność na śmierć komórki (Luo et al., 2005). W badaniu tym wykazano, że nowa funkcja apoptozy była spowodowana IkBa, podjednostką hamującą aktywację NF-kB. Pazarentzos i in., (2014) stwierdził, że IkBa lokalizuje się do OMM, gdzie wchodzi w interakcję z zależnym od napięcia kanałem anionowym (vdac) i heksokinazą mitochondrialną II (HKII), aby ustabilizować ten kompleks i zapobiec uwalnianiu cytochromu C za pośrednictwem Bax dla apoptozy. Bax jest członkiem rodziny białek Bcl-2, które okazały się regulatorami programowanej śmierci komórki (Karbowski et al., 2006).
inne badania wskazywały również na rolę NF-kB w bardziej bezpośredniej regulacji apoptozy w mitochondriach. W badaniu Liu et al., (2004), hamowanie samego NF-kB w makrofagach spowodowało uwalnianie cytochromu c. Przypomnijmy, że cytochrom c jest odpowiedzialny za przenoszenie elektronów z kompleksu III do kompleksu IV i że uwalnianie cytochromu C do cytoplazmy, aktywatora kaspaz, jest kluczowym krokiem w wyzwalaniu apoptozy.
NF-kB i oddychanie mitochondrialne
czynnik jądrowy kappa B został wykazany w wielu badaniach w celu promowania tumorigenezy. To, jak to się dzieje, nie było do końca jasne. W przełomowym badaniu Mauro et al., (2011), stwierdzono, że NF-kB reguluje oddychanie mitochondrialne w komórkach raka jelita grubego. Tutaj ustalono, że ta funkcja NF-kB tłumi efekt Warburga. Przypomnijmy, że efekt Warburga (Vander Heiden et al., 2009) opisuje obserwację, że komórki nowotworowe mają tendencję do faworyzowania metabolizmu przez glikolizę, a nie przez bardziej efektywny szlak fosforylacji oksydacyjnej. Tak więc w tym badaniu autorzy stwierdzili, że NF-kB organizuje sieci metabolizmu energetycznego poprzez kontrolowanie równowagi między wykorzystaniem glikolizy a oddychaniem mitochondrialnym., Co ciekawe, odkryli rolę NF-kB w adaptacji metabolicznej w normalnych komórkach i w raku. Ich wyniki sugerowały ponadto, że hamowanie metabolizmu mitochondrialnego w ustalonych komórkach nowotworowych poprzez hamowanie NF-kB i metforminy zmniejsza guzy.
NF-kB i ekspresja genów mitochondrialnych
czynnik jądrowy kappa B jest znanym regulatorem ekspresji genów – zarówno negatywnie, jak i pozytywnie (Mattson et al., 2000). Jednak sposób, w jaki NF-kB reguluje lub wpływa na ekspresję mitochondrialnego genu kodowanego nuklearnie, jest mniej zrozumiały., Ludzka mtDNA posiada 37 genów kodujących 13 polipeptydów. Wykazano, że geny mtDNA kodują wiele podjednostek wszystkich 5 kompleksów łańcucha transportu elektronów (ETC), 2 rRNA i 22 Trna. Aczkolwiek, większość podjednostek ETC są kodowane przez DNA jądrowego, które mogą być pod wpływem aktywności NF-kB (Calvo et al., 2016).
na przykład twierdzono (Cogswell et al., 2003), że szlak NF-kB może negatywnie regulować ekspresję genu mitochondrialnego związanego z podjednostką COX III., Podjednostka COX III jest kodowana przez mtDNA i jest składnikiem kompleksu IV w mitochondriach. Działa jako podjednostka katalityczna w kompleksie IV, który jest kompleksem związanym z mitochondrialnym zużyciem tlenu. W badaniu przeprowadzonym przez Cogswell et al. (2003), modulacja aktywacji NF-kB spowodowała utratę ekspresji zarówno mRNA Cox III, jak i mRNA cytochromu B. Inne badania potwierdzają rolę NF-kB regulującego dodatkowe geny mitochondrialne, takie jak Cox I I Cytb (Psarra and Sekeris, 2008, 2009; Barshad et al., 2018b)., Dodatkowo podjednostka NF-kB p65 obniżyła poziom kodowanego mtDNA mRNA cytb, prawdopodobnie przez wiązanie się z pętlą D w komórkach ludzkich przy braku p53 (Johnson et al., 2011). Ogólnie wyniki te sugerują, że sygnalizacja NF-kB może wpływać na aktywność enzymatyczną kompleksów oddechowych itp.
NF-kB pośredniczy w dysfunkcji wywołanej przez Aß w mitochondriach
choroba Alzheimera (AD) jest związana z gromadzeniem się blaszek Aß i/lub pojawieniem się splątań neurofibrilarnych (NFTS) w niektórych regionach mózgu (Duyckaerts et al., 2009)., Istnieją jednak kontrowersje wokół tego, czy Aß jest czynnikiem sprawczym reklamy lub czy Aß jest po prostu skorelowany ze starzeniem się. Gromadzenie dowodów (Aliev et al., 2009; Correia et al., 2012; Cadonic et al., 2016; Cardoso et al., 2017; Djordjevic et al., 2017) również teraz wskazuje na zmiany w metabolizmie mózgu napędzane przez dysfunkcję mitochondriów jako proces centralny do wielu związanych z wiekiem zaburzeń neurodegeneracyjnych, w tym AD. Dodając do tego dowody, istnieją również zaburzenia aktywności enzymatycznej kompleksów białkowych ETC i zmiany w aktywności enzymatycznej przeciwutleniaczy (Kolosova et al.,, 2017) w AD. W szczególności wykazano negatywny wpływ na aktywność kompleksu IV w AD (Mutisya et al., 1994).
w ostatnim badaniu Shi i wsp. (2014), stwierdzono, że Aß upośledza funkcję mitochondriów poprzez sygnalizację NF-kB. Co więcej, Shi et al. (2014) wykazał tutaj, że Aß zmniejsza ekspresję podjednostki COX III poprzez szlak NF-kB., Co ważne, aby wyeliminować możliwość fosforylacji IkBa przez Aß w cytoplazmie (a następnie przetransportowania do mitochondriów), izolowane mitochondria inkubowano z Aß w obecności (lub braku) blokera NF-kB, czyli BAY11-7082. Tutaj odkryli aß indukowane fosforylacji i degradacji IkBa w izolowanych mitochondriach.
wyniki te mają również istotne implikacje dla leczenia AD, jak wykazały ostatnie badania przeprowadzone przez Snow et al. (2018) i jak wykazano w innych powiązanych badaniach (Djordjevic et al., 2017; Adlimoghaddam et al.,, 2019), które sugerują, że sygnalizacja NF-kB w mitochondriach może mieć wartość terapeutyczną. Na przykład w śniegu i in.wykazano, że kreatyna – znany modulator funkcji mitochondrialnej (Tarnopolsky and Beal, 2001) zwiększa i pozytywnie zmienia poziom białka podjednostek CaMKII, PSD-95 I Complex 1 u myszy karmionych kreatyną, podczas gdy podjednostka IkB hamująca NF-kB została zmniejszona. Dodatkowe informacje na temat potencjalnego działania terapeutycznego kreatyny na czynność mitochondrialną oraz w zaburzeniach mitochondrialnych lub innych zaburzeniach neurologicznych można znaleźć w badaniach i recenzjach Matthews et al., (1998), Klivenyi et al. (1999), Tarnopolsky and Beal (2001), Hersch et al. (2006), Rodriguez et al. (2007) i Beal (2011).
rola NF-kB w stanach zapalnych i metabolizmie mitochondrialnym
, 2003; Mauro et al., 2011; Moretti et al., 2012) sugerują, że sygnalizacja NF-kB, która jest mediatorem procesów zapalnych, funkcjonuje również jako regulator i integrator metabolizmu energetycznego. W ostatnim badaniu Zhong et al. (2016) wykazano, że NF-kB ogranicza aktywację inflammasome poprzez eliminację uszkodzonych mitochondriów., Zaskakująco, NF-kB wydawało się, że zarówno prime nlrp-3 inflammasome do aktywacji, jak i zapobiegał nadmiernemu zapaleniu i powstrzymywał aktywację inflammasome NLRP – 3; chociaż mechanizm ograniczania był słabo zdefiniowany. Tutaj spekulowano, że oprócz NF-kB jest aktywatorem genów zapalnych, funkcjonował również w tym badaniu poprzez ograniczenie aktywacji zapalnej NLRP3 i produkcji IL-1β. Ponadto stwierdzono, że indukcja p62 była odpowiedzialna za aktywność hamującą inflammasome przez NF-kB., Wydaje się, że NF-kB może powstrzymać własny stan zapalny w makrofagach poprzez promowanie usuwania uszkodzonych mitochondriów (mitofagii) za pośrednictwem P62 po interakcji makrofagów z różnymi aktywatorami zapalnymi NLRP3.
wnioski
ponad 10 lat temu wykryto NF-kB w mitochondriach. Co zaskakujące, dla tak ważnego czynnika transkrypcyjnego poczyniono niewielkie postępy w odkrywaniu specyficznych ról NF-kB wpływających na mitochondrion., Niektóre badania, jak opisano powyżej, dostarczają dowodów na NF-kB w dynamice mitochondriów, apoptozie, kontroli oddechowej, ekspresji genów i mechanizmach choroby (ryc. 1). Jednak powielanie tych wyników i ogólna Walidacja są nadal konieczne przez inne laboratoria. Niektóre dodatkowe spostrzeżenia można wyciągnąć z faktu, że inne czynniki transkrypcyjne mające wpływ na geny jądrowe, takie jak AP-1, p53, CREB, C-Myc, Wnt13, Dok-4, HMGA1 i C-Src zostały również wykryte w mitochondriach (Psarra and Sekeris, 2008)., Co ciekawe, miejsca wiązania w genomie mitochondrialnym (homologicznym do miejsc wiązania w DNA jądrowym) dla niektórych z tych czynników zostały określone (Psarra and Sekeris, 2008), gdzie podejrzewa się rolę transkrypcji mitochondrialnej i apoptozy i wykazują pewne ogólne wzorce aktywności. Na przykład można argumentować, że niektóre z tych czynników (NF-kB, CREB i AP-1) wiążą się z genomami mitochondrialnymi i głównie osłabiają ekspresję genów mitochondrialnych (Blumberg et al., 2014), mając jednocześnie stymulujący wpływ na transkrypcję genów jądrowych., Jednak wyraźnie potrzeba więcej pracy, aby nie tylko znaleźć dokładne role aktywności, ale także określić, czy ogólne wzorce aktywności naprawdę istnieją.
Po zapoznaniu się z tą literaturą staje się również jasne, że rola NF-kB w regulacji oddychania mitochondrialnego ma głębokie implikacje i wykazuje poziom złożoności, który wcześniej nie był doceniany. Na przykład Mauro et al. (2011) dane ustalają rolę NF-kB w adaptacji metabolicznej w normalnych komórkach i w raku, a także sugerują konsekwencje dla innych stanów chorobowych, takich jak AD., Ponadto, biorąc pod uwagę, że NF-kB może powstrzymać własny stan zapalny, jak wykazały Zhong et al. (2016), nie tylko jest zaskakujące, ale dodatkowo ilustruje złożoność sygnalizacji NF-kB w funkcji mitochondrialnej.
w niniejszym przeglądzie przeprowadzono badania nad rolą NF-kB w funkcji mitochondriów i wydaje się, że badania w tym obszarze rosną. Komplikowanie wyników jest jednak obserwacja, że wiele czynników odgrywa podobną rolę w funkcji mitochondriów i dlatego konieczne są szczegółowe badania specyficzne dla każdego czynnika., Podsumowując, możemy ponownie Zadać pytanie – Co NF-kB robi w I do mitochondrionu? Natychmiastowa i skrócona odpowiedź byłaby-dużo!
wkład autora
autor stworzył temat i napisał rękopis.
finansowanie
praca ta była wspierana przez fundusze z Canadian Institutes of Health Research (CIHR), Canadian Agricultural Partnership (CAP), St.Boniface Hospital Research Foundation, Alzheimer Society of Manitoba, Research Manitoba, Szanowny Douglas i Patricia Everett oraz Royal Canadian Properties Limited Endowment Fund.,
Oświadczenie o konflikcie interesów
autor oświadcza, że badanie zostało przeprowadzone w przypadku braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
podziękowania
autor chciałby podziękować Dr Grant Hatch za recenzję manuskryptu.
Sygnalizacja do NF-kappaB. Geny Dev. 18, 2195–2224.
PubMed Abstract/Google Scholar
Wang, X. and Chen, X. J. (2015)., Sieć cytozolowa hamująca stres proteostatyczny w mitochondriach i śmierć komórek. Przyroda 524, 481-484. doi: 10.1038 / nature14859
PubMed Abstract | CrossRef Full Text | Google Scholar