wprowadzenie
ukończenie projektu human genome było ważnym kamieniem milowym dla wiedzy medycznej, dostarczając informacji niezbędnych do zrozumienia unikalnych cech każdego osobnika.1 logiczną konsekwencją tej wiedzy byłaby możliwość zastosowania konkretnych testów diagnostycznych i zabiegów u każdego pacjenta w oparciu o jego indywidualną informację genetyczną. Ta nowa forma opieki medycznej nazywa się medycyną spersonalizowaną.,2 jednak, pomimo wielkiego postępu, który doprowadził do poznania ludzkiego genomu, jego tłumaczenie do diagnostycznych i spersonalizowanych metod leczenia było mniej niż oczekiwano. Obecnie podejmowane są w tym kierunku dwie główne inicjatywy: Biologia systemowa3 i farmakogenetyka.,4 ostatecznym celem tych inicjatyw jest opracowanie praktyki medycznej dostosowanej do cech każdej osoby, która może przewidzieć początek lub przebieg danej choroby, umożliwić opracowanie odpowiednich strategii zapobiegania i wreszcie umożliwić pacjentowi udział w podejmowaniu decyzji. Lek ten został nazwany lekiem P4.5
Mukowiscydoza (CF) pozostaje najczęstszą i śmiertelną chorobą genetyczną wśród osób rasy kaukaskiej., Występuje on w tempie 1 na 2500-6000 noworodków, w zależności od regionu i pochodzenia etnicznego, oraz w odsetku zdrowych nosicieli, zmieniającym się między 1:20 a 37,6 w Hiszpanii, dzięki stopniowemu wprowadzaniu programów badań przesiewowych noworodków w różnych społecznościach, obserwuje się mniejszą częstość występowania CF niż wcześniej szacowano w 2009 r., tj. 1/5840 w Katalonii i 1/6602 na Balearach.7 szacuje się, że na całym świecie jest 70000 chorych na CF.,8 choroba jest spowodowana mutacjami w genie kodującym regulator przewodzenia białka torbielowatego (CFTR), kanał chlorkowy biorący udział w uwalnianiu adenozynotrójfosforanu i regulacji innych kanałów transportu jonów. Białko to ulega ekspresji w komórkach nabłonka oddechowego, trzustce, drogach żółciowych, gruczołach potowych i układzie moczowo-płciowym., Jego zmiana prowadzi do nieprawidłowości w transporcie jonów, tak że pacjenci wytwarzają gęsty, lepki śluz, który zatyka kanały narządu, w którym się znajduje, a więc zmiana prezentuje efekty wielosystemiczne, które określają szeroki zakres objawów klinicznych CF. Pomimo znacznych postępów w leczeniu mukowiscydozy, które doprowadziły do dłuższego przeżycia (obecna mediana szacowana jest na 37,5 roku) 9, wciąż pozostaje wiele do zrobienia, aby zapewnić pacjentom z mukowiscydozą podobną ilość i jakość życia jak u osób bez choroby., W tym kontekście konieczne są nowe metody leczenia mające na celu zmniejszenie zachorowalności i zwiększenie przeżywalności.
CF jest przykładem choroby dobrze przygotowanej do skorzystania z medycyny spersonalizowanej. Z jednej strony jest to choroba monogenna, spowodowana mutacjami w określonym Genie. Patofizjologia podmiotu jest dobrze scharakteryzowana, a cele terapeutyczne są jasne. Ponadto diagnoza choroby wymaga badań genetycznych w celu identyfikacji rodzaju choroby, więc dokładna wada genetyczna jest określana w każdym przypadku.,10
obecnie dwa bardzo różne podejścia są ukierunkowane na skorygowanie podstawowej wady: terapia genowa, mająca na celu skorygowanie zmiany genetycznej, i terapia cząsteczkowa, mająca na celu skorygowanie wady funkcjonalnej na poziomie białka. Celem terapii genowej jest wprowadzenie prawidłowych kopii genów w drogach oddechowych pacjentów z mukowiscydozą. Polega ona na wstawieniu rekombinowanego wektora wirusa, którego DNA zostało wyekstrahowane i zastąpione przez nowe DNA terapeutyczne. Wektor wirusa służy jako nośnik do wprowadzania nowego DNA do komórki docelowej., Do tej pory stosowano różne rodzaje wirusów, takie jak adenowirus czy lentivirus. Ponadto, nie wirusowe cząstki, takie jak nanocząstki zdolne do wstawiania DNA, również zostały opracowane.Jednak dotychczasowe wyniki były słabe, ponieważ czas ekspresji wprowadzonego genu był krótki dla obu typów wektorów.12 Brytyjskie Konsorcjum terapii genowej opracowuje II Fazę badania klinicznego w celu oceny skuteczności klinicznej zoptymalizowanego wektora plazmidowego / liposomalnego DNA.13 celem rekrutacji jest 130 pacjentów, a wyniki spodziewane są w 2014 r. (NCT01621867).,Z drugiej strony, terapia ukierunkowana na przywrócenie funkcji białka CFTR była bardziej skuteczna. W ostatnich latach, wyniki zaczynają przyjść na leki, które działają bezpośrednio na białko CFTR. W rzeczywistości w styczniu 2012 roku pierwszy lek do korygowania wad mutacji Gly551Asp został wprowadzony do obrotu w USA. W poniższych sekcjach dokonamy przeglądu dostępnych informacji na temat postępu medycyny spersonalizowanej dla mukowiscydozy oraz dostępnych zabiegów mających na celu skorygowanie wady powodującej chorobę na poziomie białka., W niniejszym przeglądzie zostanie użyta nomenklatura używana do opisu mutacji genów CFTR opracowanych przez Towarzystwo zmienności genomu człowieka (Human Genome Variation Society15).
mutacje i defekt białka
CF jest chorobą dziedziczną autosomalnie recesywną, więc mutacja musi być obecna w obu kopiach genu CFTR, aby mogła zostać dotknięta. Do tej pory zidentyfikowano ponad 1900 mutacji genu CFTR związanych z chorobą w sekwencji kodującej, messenger RNA lub innych elementach. Mutacje w genie CFTR są dostępne do konsultacji w bazie danych mutacji mukowiscydozy.,16 pierwszą opisaną mutacją i najczęstszą na świecie jest Phe508del, ale istnieją inne specyficzne mutacje z różną częstotliwością wśród różnych grup etnicznych. W Hiszpanii średnia częstość mutacji Phe508del wynosi od 50% do 60% wszystkich badanych chromosomów, drugi najczęściej jest Gly542X z 4% -8%, a następnie Asn1303Lys w 2% -4% przypadków. Dotychczas opisane mutacje dzieli się na sześć typów lub klas w zależności od mechanizmu powodującego chorobę.17 te typy mutacji są podsumowane na Rys. 1., Mutacje klasy I prowadzą do przedwczesnego zatrzymania kodonu w przekaźniku RNA, co uniemożliwia translację całego białka. Tak więc produkowane białko jest krótkie i niefunkcjonujące. Mutacje klasy II kodują strukturalnie nieprawidłowe i błędnie sfałdowane białko, które jest usuwane przez retikulum endoplazmatyczne przed dotarciem do powierzchni komórki. Najczęstsza mutacja w CF, Phe508del, należy do tej grupy. W przypadku mutacji w klasach III-VI białka docierają do powierzchni komórki, ale nie działają prawidłowo. Mutacje klasy III powodują zmniejszoną aktywację kanału, więc kanały pozostają zamknięte., Mutacje klasy IV powodują spadek przewodności jonów przez kanał. Mutacje Klasy V kodują niewielkie białka, powodując zmniejszenie ilości CFTR na powierzchni komórki, tak że zachodzi pewna funkcja, ale na obniżonym poziomie. W końcu mutacje klasy VI prowadzą do skrócenia okresu półtrwania z powodu niestabilności białek i mogą również uszkodzić regulację sąsiednich kanałów CFTR na powierzchni komórki.
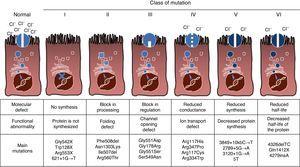
rodzaje mutacji w mukowiscydozie.
modulatory CFTR
trzy główne klasy zostały zidentyfikowane w rozwoju leków do naprawy białka CFTR.11 pierwsza grupa to przedwczesne hamowanie kodonów (mutacje klasy I). Leki te zapobiegają identyfikacji tego przedwczesnego kodonu zatrzymania, dzięki czemu synteza białek może trwać do zakończenia. Druga grupa to korektory CFTR. Związki te mają za zadanie korygować wady transportu złożonego białka (mutacje klasy II) do błony komórkowej, gdzie może ono funkcjonować prawie normalnie., Trzecią grupę stanowią tzw. potencjatory CFTR. Są to leki przeznaczone do celowania w białko CFTR na powierzchni komórki w celu poprawy jego funkcji. Tak więc potencjatory te mogą działać na mutacje klasy III, IV, V i VI. Obecnie badane są liczne cząsteczki wykorzystujące te różne mechanizmy, z których jeden już trafił na rynek: iwakaftor (VX-770) jest potencjatorem CFTR zatwierdzonym w USA w styczniu 2012 roku do leczenia pacjentów z mukowiscydozą w wieku powyżej 6 lat z mutacją Gly551Asp.,
leczenie mutacjami klasy I
Około 10% przypadków CF jest spowodowanych mutacjami klasy I. Pierwszymi lekami stosowanymi w tej klasie były aminoglikozydy. Kilka lat temu donoszono, że gentamycyna ma zdolność maskowania przedwczesnego kodonu stop zapobiegającego syntezie białka CFTR. Osiąga się to poprzez wstawienie aminokwasu, który umożliwia rybosom kontynuowanie czytania genu, produkując pełnowymiarowe białko. Badania przedkliniczne wykazały, że białko można zsyntetyzować w modelu mysim, a 35% funkcji białka odzyskano in vitro.,Wpływ dożylnego podawania gentamycyny oceniano w dwóch badaniach z udziałem pacjentów z mukowiscydozą z różnymi typami mutacji klasy I. Jeden został przeprowadzony w USA na pięciu pacjentach, 20, a drugi na 18 pacjentów we Francji.Jednakże, chociaż odpowiedź była pozytywna, wyniki były bardzo zróżnicowane, więc korzyści nie były powszechne. Ponadto problemy z toksycznością aminoglikozydów przyczyniły się do niekorzystnego profilu.
syntetyczną alternatywą jest ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., Jest to cząsteczka zaprojektowana, aby umożliwić rybosomom odczytanie informacji genetycznej podczas „pomijania” przedwczesnego kodonu stop, produkującego funkcjonalne białko CFTR.Farmakokinetykę produktu ataluren wykazano w modelach zwierzęcych oraz w badaniach II Fazy. W dwóch małych badaniach wstępnych, 23,24 grupa pacjentów z mukowiscydozą leczonych doustnym preparatem ataluren wykazała poprawę zaburzeń elektrofizjologicznych choroby, ze wzrostem liczby komórek w nosie wyrażających białko na ich powierzchni., Następnie w innym małym badaniu z udziałem 19 pacjentów oceniano różne dawki produktu ataluren podawanego doustnie co 8 godzin, z poprawą aktywności CFTR i parametrów klinicznych oraz dobrym profilem bezpieczeństwa.25 wyniki badania klinicznego III fazy ataluren, które nie zostały jeszcze oficjalnie opublikowane, zostały przedstawione na konferencji północnoamerykańskiej w 2012 r. Łącznie 238 pacjentów w wieku powyżej 6 lat losowo przydzielono do grupy otrzymującej ataluren 10, 10, 20 mg/kg mc. lub placebo co 8 godzin przez 48 tygodni., Po 48 tygodniach leczenia nie stwierdzono istotnych różnic w FEV1 w grupie ataluren w porównaniu z placebo (-2, 5% w grupie ataluren w porównaniu z -5, 5% w grupie placebo, P=NS). Po stratyfikacji przez przewlekłe stosowanie nebulizowanych antybiotyków różnica w średniej zmianie po 48 tygodniach wynosiła 6,7% na korzyść produktu ataluren u pacjentów nie leczonych tobramycyną, podczas gdy nie było różnicy w zmianie FEV1 u pacjentów otrzymujących nebulizowaną tobramycynę. Coś podobnego wystąpiło z odsetkiem zaostrzeń, gdzie różnica między obiema grupami była znacząca., Po stratyfikacji u pacjentów z grupy ataluren nie leczonych nebulizowaną tobramycyną stwierdzono procentowy spadek zaostrzeń o 43% w porównaniu z grupą placebo. Badania potencjału nosa i chlorku potu nie wykazały różnic między grupami, niezależnie od stosowania nebulizowanych antybiotyków. Autorzy doszli do wniosku, że korzyści były większe u pacjentów nie otrzymujących przewlekłej antybiotykoterapii z nebulizowanym aminoglikozydem, spekulując, że tobramycyna i ataluren oddziaływały na poziomie rybosomalnym, powodując antagonizm, gdy są stosowane jednocześnie.,26
leczenie mutacjami klasy II
mutacje klasy II, będące najczęstszą mutacją choroby (Phe508del), są obecne u dużej liczby pacjentów z mukowiscydozą, co czyni je głównym celem badań nad mukowiscydozą. Badano różne cząsteczki, z których większość została opracowana przez Vertex Pharmaceuticals Inc. Pierwszy związek korektora, lumakaftor (VX-809), wykazał dobrą skuteczność in vitro, poprawiając transport chlorków w 14%.,Jednak wyniki były nieco rozczarowujące u pacjentów, ponieważ poprawa stężenia chlorków w pocie była bardzo mała (7 mmol/l) i nie stwierdzono zmian potencjału nosowego.Działanie iwakaftora CFTR (VX-770), leku występującego w mutacjach III klasy (patrz opis poniżej), badano również u pacjentów homozygotycznych dla Phe508del. Wyniki badania DISCOVER pokazują, że iwakaftor nie jest związany ze znaczną poprawą FEV1, jakością życia ani liczbą zaostrzeń w porównaniu z placebo.,29
ponieważ lumakaftor może wspomagać dostarczanie Phe508del-CFTR do powierzchni komórki i iwakaftor zwiększa czas otwarcia i przewodzenie chlorków przez komórkę nabłonkową, poprawa podstawowej wady Phe508del może być możliwa dzięki kombinacji obu cząsteczek. Badania in vitro z połączonym iwakaftorem i lumakaftorem w nabłonku oddechowym z mutacją Phe508del wykazały, że lumakaftor zwiększa transport chlorków CFTR o 15% w monoterapii, a po dodaniu iwakaftoru transport wzrasta do prawie 30%., To połączenie leków było badane w badaniu II fazy u pacjentów z mutacją Phe508del. Pełne wyniki nie zostały jeszcze opublikowane, ale wstępne dane sugerują korzystny wpływ na czynność płuc w homozygotycznym Phe508del, ale nie w heterozygotyczności.Opracowywane są dwa badania kliniczne z udziałem pacjentów w wieku 12 lat lub starszych homozygotycznych dla mutacji Phe508del w celu oceny skojarzonego iwakaftoru i lumakaftoru; są to badania ruchu (NCT01807923)31 i transportu (nct01807949)32. Wyniki mogą pozwolić około połowie populacji CF na leczenie modulujące CFTR., Konieczne są jednak badania w celu oceny wpływu terapii skojarzonej u heterozygotycznych osób Phe508del.
inną alternatywą jest związek korektora VX-661, a badania są obecnie w toku. Jego skuteczność jest testowana samodzielnie i w połączeniu z iwakaftorem (VX-770), a wyniki będą wkrótce dostępne (NCT01531673).
iwakaftor (VX-770) jest potencjatorem CFTR, który moduluje funkcję nieprawidłowego białka.,Cząsteczka ta została pierwotnie zaprojektowana w celu wzmocnienia funkcji CFTR w hodowlach komórek nabłonka oddechowego posiadających pojedynczą mutację Gly551Asp.Jest to pierwszy lek zatwierdzony w USA i Europie do leczenia mukowiscydozy u pacjentów z mutacją Gly551Asp. Po wykazaniu in vitro zdolności iwakaftoru do poprawy transportu chlorków przez błonę komórkową, przeprowadzono pierwsze badania II fazy z udziałem 39 pacjentów.W tym badaniu funkcja białka CFTR poprawiła się trzy dni po rozpoczęciu leczenia, osiągając stężenie chlorków w pocie do normalnego poziomu., W wyniku tych wyników przeprowadzono dwa dalsze badania kliniczne: badanie STRIVE z udziałem 144 pacjentów w wieku co najmniej 12 lat36 oraz badanie ENVISION z udziałem 52 dzieci w wieku od 6 do 11 lat.Oba badania obejmowały pacjentów z co najmniej jedną mutacją Gly551Asp i FEV1 pomiędzy 40% A 105%, którzy początkowo byli obserwowani przez okres 14 dni, a następnie randomizowani do grupy otrzymującej 150 mg doustnego iwakaftoru lub placebo dwa razy na dobę przez okres 48 tygodni., Po zakończeniu 48 tygodni leczenia pacjenci mieli możliwość kontynuowania badania otwartego metodą wzdłużną,badania PERSIST (nct01117012), 38 przez 96 tygodni.
u pacjentów w grupie otrzymującej iwakaftor stwierdzono poprawę o 10,6% w FEV1 (pierwszorzędowy punkt końcowy) począwszy od 15.dnia leczenia, która utrzymywała się podczas 48-tygodniowego badania. Ponadto obserwowano zmniejszenie stężenia chlorków w pocie (średnio: -48,7 mmol/L), poprawę jakości życia, zmniejszenie zaostrzeń o 55% i zwiększenie masy ciała o 2,7 kg.,
wyniki badania ENVISION pokrywają się w dużej mierze z wynikami badania u młodzieży i dorosłych, z tą różnicą, że jakość życia nie osiągnęła statystycznej różnicy. Do działań niepożądanych częściej obserwowanych w grupie leczonej zarówno w badaniach exhaust, jak i ENVISION należały infekcje górnych dróg oddechowych, przekrwienie błony śluzowej nosa, ból gardła, zawroty głowy i wysypka skórna., Wstępne wyniki po pierwszych 12 tygodniach badania PERSIST ujawniają, że poprawa czynności płuc (FEV1), objawów ze strony układu oddechowego i przyrostu masy ciała u pacjentów leczonych iwakaftorem utrzymuje się w tym okresie. Ponadto w podgrupie pacjentów, którzy przed rozpoczęciem leczenia przeszli z placebo na iwakaftor, stwierdzono poprawę FEV1 o 10,8% po 15 dniach i o 13% po 12 tygodniach, wraz ze zmniejszeniem zaostrzeń.,Pomimo dobrych wyników leczenia iwakaftorem w leczeniu mutacji Gly551Asp u dzieci w wieku powyżej 6 lat i dorosłych w wieku 48 tygodni, niektóre problemy pozostają do rozwiązania. Po pierwsze, lek nie został przetestowany u dzieci poniżej 6 lat. Jednak rozsądne wydaje się skorygowanie wady przed powstaniem nieodwracalnych uszkodzeń, biorąc pod uwagę, że zajęcie płuc rozpoczyna się przed szóstym rokiem życia. Badanie w tym przedziale wiekowym jest obecnie w toku (NCT01705145).40 Po Drugie, inną alternatywą byłoby wypróbowanie również innych mutacji klasy III., W związku z tym, badania in vitro na dziewięciu innych mutacjach wykazały bardzo podobne wyniki,41 więc uzasadnione jest oczekiwanie podobnych wyników u pacjentów. Trwa badanie kliniczne III fazy u pacjentów w wieku powyżej 6 lat z innymi mutacjami III klasy (badania KONTINUE i Konnection; nct01614470).42,43%, W tym zakresie prowadzone jest badanie kliniczne oceniające skuteczność iwakaftoru w mutacji IV klasy Arg117His (badanie KONDUCT; nct01614457).Na koniec, skuteczność i długoterminowe bezpieczeństwo stosowania iwakaftoru w okresie dłuższym niż 48 tygodni nie zostały jeszcze ustalone. Badanie obserwacyjne G551D (GOAL; NCT01521338)45 jest badaniem obserwacyjnym obejmującym pacjentów w wieku powyżej 6 lat otrzymujących iwakaftor., Ma na celu zgłaszanie skuteczności i bezpieczeństwa długotrwałego iwakaftoru, wraz z innymi wynikami zainteresowania, które obejmują mediatory zapalne w plwocinie, klirens śluzowo-żółciowy i pH przewodu pokarmowego. wyniki są spodziewane pod koniec 2013 roku.
wnioski
CF jest przykładem choroby dobrze przygotowanej do skorzystania z medycyny spersonalizowanej. Obecnie dwa bardzo różne podejścia mają na celu skorygowanie podstawowej wady: terapia genowa mająca na celu skorygowanie zmiany genetycznej oraz terapia cząsteczkami mająca na celu skorygowanie wady funkcjonalnej na poziomie białka., Ten ostatni zaczyna wykazywać obiecujące wyniki dla różnych cząsteczek w rozwoju, a jedna z nich (iwakaftor) jest już sprzedawana dla mutacji Gly551Asp klasy III, z doskonałymi wynikami u dzieci w wieku powyżej 6 lat, młodzieży i dorosłych. Ostatecznym celem jest zapewnienie leczenia korekcyjnego i wzmacniającego dla wszystkich pacjentów z mukowiscydozą bez względu na ich mutację. Ponieważ wyniki tych i innych nowych cząsteczek pojawiają się, jest prawdopodobne, że konkretna cząsteczka lub kombinacja będzie potrzebna dla każdego pacjenta, w zależności od ich istniejących mutacji., W każdym razie przyszłość jest obiecująca, a ważne kroki zostały podjęte w kierunku uzyskania leczenia, które będzie skutecznie działać na przyczynę tej choroby.
konflikty interesów
autorzy nie deklarują konfliktu interesów.