Inleiding
de voltooiing van het human genome project was een relevante mijlpaal voor medische kennis, die de informatie verschafte die nodig was om de unieke kenmerken van elk individu te begrijpen.1 de logische consequentie van deze kennis zou zijn om specifieke diagnostische tests en behandelingen toe te passen op elke patiënt op basis van hun individuele genetische informatie. Deze nieuwe vorm van medische zorg heet gepersonaliseerde geneeskunde.,2 maar ondanks de grote vooruitgang die heeft geleid tot de kennis van het menselijk genoom, is de vertaling ervan in diagnostische en gepersonaliseerde behandelingen minder dan verwacht. Op dit moment wordt in deze richting gewerkt met twee belangrijke initiatieven: systeembiologie3 en farmacogenetica.,4 het uiteindelijke doel van deze initiatieven is het ontwikkelen van een medische praktijk aangepast aan de kenmerken van elk individu die het begin of het verloop van een bepaalde ziekte kan voorspellen, het mogelijk maken passende preventiestrategieën vast te stellen en, ten slotte, de patiënt in staat te stellen deel te nemen aan de besluitvorming. Dit wordt P4-geneesmiddel genoemd.
cystische fibrose (CF) blijft de meest voorkomende en dodelijke genetische ziekte onder Kaukasiërs., Het komt voor in een tempo van 1 op 2500-6000 pasgeborenen, afhankelijk van de regio en etnische afkomst, en in een aantal gezonde dragers, variërend tussen 1:20 en 37,6 in Spanje, dankzij de geleidelijke invoering van neonatale screeningsprogramma ‘ s in de verschillende gemeenschappen, wordt nu een lagere incidentie van CF gezien dan eerder werd geschat in 2009, namelijk 1/4430 levendgeborenen in Galicië, 1/4339 in Castilla-Leon, 1/5376 in Murcia, 1/5840 in Catalonië en 1/6602 op de Balearen.7 naar schatting zijn er wereldwijd 70000 CF-patiënten.,8 de ziekte wordt veroorzaakt door mutaties in het gen dat codeert voor de regulerende eiwit cystic fibrosis transmembrane conductance regulator (CFTR), een chloride kanaal betrokken bij de afgifte van adenosine trifosfaat en regulering van andere ionentransportkanalen. Dit eiwit wordt uitgedrukt in epitheliale cellen van de luchtwegen, pancreas, galwegen, zweetklieren en urogenitaal systeem., De verandering leidt tot een afwijking in ionentransport, zodat patiënten dik, kleverig slijm produceren dat de kanalen van het orgaan waar het zich bevindt verstopt en zo de verandering multisystemische effecten vertoont die het brede scala van klinische manifestaties van CF bepalen. Ondanks grote vooruitgang in de behandeling van CF die heeft geresulteerd in een langere overleving (huidige mediaan wordt geschat in 37,5 jaar) 9,is er nog een lange weg te gaan om ervoor te zorgen dat patiënten met CF een hoeveelheid en kwaliteit van leven hebben die vergelijkbaar is met die van patiënten zonder de ziekte., In deze context zijn nieuwe behandelingen nodig om de morbiditeit te verminderen en de overleving te verhogen.
CF is een voorbeeld van een ziekte die goed gepositioneerd is om gebruik te maken van gepersonaliseerde geneeskunde. Aan de ene kant is het een monogene ziekte, veroorzaakt door de mutaties in een specifiek gen. De pathofysiologie van de entiteit is goed gekarakteriseerd en de therapeutische doelen zijn duidelijk. Voorts vereist de diagnose van de ziekte genetische tests voor de identificatie van het ziektetype, zodat het exacte genetische defect in elk geval wordt bepaald.,10
Momenteel zijn twee zeer verschillende benaderingen gericht op het corrigeren van het basisdefect: gentherapie, gericht op het corrigeren van de genetische verandering, en molecuultherapie, gericht op het corrigeren van het functionele defect op het eiwitniveau. De focus van gentherapie is de introductie van normale gen kopieën in de luchtwegen van CF patiënten. Het gaat om de toevoeging van een recombinante virale vector, waarvan het DNA is geëxtraheerd en vervangen door het nieuwe therapeutische DNA. De virale vector dient als voertuig voor het invoegen van het nieuwe DNA in de doelcel., Diverse types van virussen, zoals adenovirus of lentivirus, zijn tot op heden gebruikt. Voorts zijn niet-virale deeltjes, zoals nanoparticles geschikt om DNA in te voegen, ook ontwikkeld.11 nochtans, zijn de resultaten tot nu toe slecht geweest, omdat de duur van uitdrukking van het geïntroduceerde gen met beide types van vectoren kort was.12 Het Britse Consortium voor gentherapie ontwikkelt een Fase II klinisch onderzoek om de klinische effectiviteit van een geoptimaliseerde plasmidische/liposomale DNA-vector te evalueren.13 het wervingsdoel is 130 patiënten en de resultaten worden verwacht in 2014 (NCT01621867).,
anderzijds is een therapie gericht op het herstellen van de functie van het CFTR-eiwit succesvoller geweest. In de afgelopen jaren beginnen de resultaten door te komen op geneesmiddelen die direct inwerken op het CFTR-eiwit. In feite, in januari 2012, werd het eerste medicijn voor het corrigeren van gly551asp-mutatiedefecten op de markt gebracht in de VS. In de volgende paragrafen zullen we de beschikbare informatie over de voortgang van gepersonaliseerde geneeskunde voor CF en beschikbare behandelingen bekijken die gericht zijn op het corrigeren van het defect dat de ziekte veroorzaakt op het eiwitniveau., In dit overzicht zal de nomenclatuur gebruikt worden voor de beschrijving van CFTR genmutaties ontwikkeld door de Human Genome Variation Society15.
mutaties en Eiwitdefect
CF is een autosomaal recessieve erfelijke ziekte, dus de mutatie moet aanwezig zijn in beide kopieën van het CFTR gen om te worden beïnvloed. Tot op heden zijn meer dan 1900 CFTR genmutaties geassocieerd met de ziekte geà dentificeerd in de codeeropeenvolging, boodschappersRNA of andere elementen. Mutaties in het CFTR gen zijn beschikbaar voor consultatie in de Cystic Fibrosis Mutation Database.,16 De eerste beschreven mutatie, en de meest voorkomende wereldwijd, is Phe508del, maar er zijn andere specifieke mutaties met verschillende frequentie tussen verschillende etnische groepen. In Spanje is de gemiddelde frequentie van phe508del-mutatie tussen 50% en 60% van alle onderzochte chromosomen, de tweede meest voorkomende is Gly542X met 4% -8%, gevolgd door Asn1303Lys in 2% -4% van de gevallen. De tot op heden beschreven mutaties zijn ingedeeld in zes typen of klassen volgens het mechanisme dat de ziekte veroorzaakt.17 deze soorten mutaties worden samengevat in Fig. 1., De veranderingen van klasse I leiden tot een voortijdig stopcodon in het boodschapper RNA dat Vertaling van het volledige proteã ne verhindert. Aldus, is het geproduceerde eiwit kort en niet-functioneert. Klasse II de veranderingen coderen een structureel abnormale en misfolded proteã ne die door het endoplasmic reticulum wordt verwijderd alvorens de celoppervlakte te bereiken. De meest voorkomende mutatie bij CF, Phe508del, behoort tot deze groep. In het geval van mutaties in de klassen III tot en met VI bereiken de eiwitten het celoppervlak, maar functioneren ze niet goed. Klasse III mutaties veroorzaken een verminderde kanaalactivering, zodat kanalen gesloten blijven., Klasse IV mutaties veroorzaken een afname van de iongeleiding door het kanaal. De mutaties van klasse V coderen minder belangrijke proteã nen die in een verminderde hoeveelheid CFTR in het celoppervlak resulteren, zodat een bepaalde functie, maar op een verminderd niveau voorkomt. Tenslotte leiden de veranderingen van klasse VI tot een verkorte halveringstijd toe te schrijven aan eiwitinstabiliteit en kunnen de verordening van naburige CFTR kanalen in de celoppervlakte ook beschadigen.
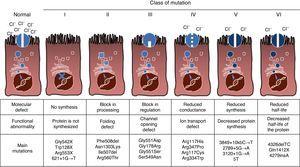
soorten mutaties bij cystische fibrose.
CFTR-modulatoren
drie hoofdklassen zijn geïdentificeerd bij de ontwikkeling van geneesmiddelen voor het herstellen van CFTR-eiwit.11 de eerste groep zijn premature stop codon suppressoren (klasse I mutaties). Deze drugs verhinderen identificatie van dit voortijdige eindcodon, zodat eiwitsynthese tot voltooiing kan doorgaan. De tweede groep zijn de CFTR-correctoren. Deze samenstellingen worden ontworpen om tekorten in het vervoer van gevouwen proteã ne (klasse II veranderingen) aan het celmembraan te corrigeren, waar het bijna normaal kan functioneren., De derde groep bestaat uit de zogenaamde CFTR potentiatoren. Dit zijn drugs die worden ontworpen om de proteã ne van CFTR op de celoppervlakte te richten om zijn functie te verbeteren. Aldus, kunnen deze potentiators op Klasse III, IV, V en VI veranderingen inwerken. Momenteel worden talrijke moleculen onderzocht die gebruik maken van deze verschillende mechanismen, waarvan er één reeds op de markt is gekomen: Ivacaftor (VX-770) is een CFTR potentiator goedgekeurd in de VS in januari 2012 voor de behandeling van CF patiënten ouder dan 6 jaar die de Gly551Asp mutatie hebben.,
Klasse I Mutatiebehandelingen
Ongeveer 10% van de gevallen van CF worden veroorzaakt door Klasse I mutaties. De eerste geneesmiddelen die voor deze klasse werden gebruikt, waren aminoglycosiden. Enkele jaren geleden werd gemeld dat gentamicine het vermogen heeft om het voortijdige stopcodon te maskeren dat de synthese van het CFTR-eiwit verhindert. Dit wordt bereikt door toevoeging van een aminozuur dat het ribosoom toestaat om verder te lezen het gen, veroorzakend een proteã ne van volledige lengte. Preklinische studies toonden aan dat het eiwit in een muismodel kon worden gesynthetiseerd en dat 35% van de eiwitfunctie in vitro werd teruggevonden.,Het effect van intraveneuze toediening van gentamicine werd beoordeeld in twee studies met CF patiënten met verschillende typen klasse I mutaties. Eén werd uitgevoerd in de VS bij vijf patiënten,20 en een andere omvatte 18 patiënten in Frankrijk.21 hoewel de respons positief was, liepen de resultaten sterk uiteen, zodat het voordeel niet universeel was. Bovendien droegen toxiciteitsproblemen met aminoglycosiden bij tot een ongunstig profiel.een synthetisch alternatief is ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., Dit is een molecuul dat wordt ontworpen om ribosomen toe te laten om de genetische informatie te lezen terwijl het “overslaan” van het voortijdige eindcodon, die een functionele proteã ne van CFTR produceren.De farmacokinetiek van ataluren is aangetoond in diermodellen en in Fase II-onderzoeken. In twee kleine voorstudies,23, 24, toonde een groep CF patiënten behandeld met orale ataluren een verbetering van de elektrofysiologische afwijkingen van de ziekte, met een toename van het aantal cellen in de neus die het eiwit op hun oppervlak tot expressie brengen., Vervolgens evalueerde een ander klein onderzoek met 19 patiënten verschillende doses ataluren die om de 8 uur oraal werden toegediend, met verbeteringen in de CFTR-activiteit en klinische parameters en een goed veiligheidsprofiel.De resultaten van een fase III klinische studie met ataluren die nog niet formeel gepubliceerd waren, werden gerapporteerd in de Noord-Amerikaanse Cystic Fibrosis Conference van 2012. In totaal werden 238 patiënten ouder dan 6 jaar gerandomiseerd naar ataluren 10, 10, 20 mg/kg of placebo om de 8 uur gedurende 48 weken., Er waren geen significante verschillen in FEV1 in ataluren versus placebo na 48 weken behandeling (-2,5% ataluren Versus -5,5% placebo, P=ns). Wanneer gestratificeerd op basis van chronisch gebruik van vernevelde antibiotica, was er een verschil van 6,7% in gemiddelde verandering na 48 weken in het voordeel van ataluren voor patiënten die niet tobramycine gebruikten, terwijl er geen verschil was in verandering in FEV1 bij degenen die vernevelde tobramycine kregen. Iets dergelijks deed zich voor met het percentage exacerbaties, waarbij het verschil tussen beide groepen significant was., Bij gestratificeerde patiënten in de ataluren-groep die geen vernevelde tobramycine kregen, vertoonden de patiënten in de groep die geen vernevelde tobramycine kregen een procentuele afname van exacerbaties van 43% in vergelijking met de placebogroep. Nasale potentiaalverschil en zweetchloridetests toonden geen verschil aan tussen de groepen, ongeacht het gebruik van vernevelde antibiotica. De auteurs concludeerden dat de voordelen groter waren bij patiënten die geen chronische antibioticumtherapie met vernevelde aminoglycoside kregen, en speculeerden dat tobramycine en ataluren een interactie hadden op ribosomaal niveau, wat antagonisme veroorzaakte bij gelijktijdig gebruik.,
klasse II Mutatiebehandelingen
klasse II mutaties, de meest voorkomende mutatie van de ziekte (Phe508del), zijn aanwezig bij een groot aantal CF patiënten, waardoor ze een primair doelwit zijn in CF onderzoek. Verschillende moleculen zijn bestudeerd, waarvan de meeste zijn ontwikkeld door Vertex Pharmaceuticals Inc. De eerste correctorverbinding, lumacaftor (VX-809), toonde een goede in vitro werkzaamheid, waardoor het chloridetransport in 14% werd verbeterd.,De resultaten waren echter enigszins teleurstellend bij patiënten, omdat de verbetering van de chlorideconcentratie in zweet zeer klein was (7 mmol/l) en er geen veranderingen waren in de nasale potentie.
de effecten van de CFTR potentiator ivacaftor (VX-770), een geneesmiddel voor klasse III mutaties (zie beschrijving hieronder), zijn ook onderzocht bij patiënten die homozygoot waren voor Phe508del. De resultaten van de DISCOVER-studie tonen aan dat ivacaftor niet in verband wordt gebracht met een significante verbetering van FEV1, de kwaliteit van leven of het aantal exacerbaties ten opzichte van placebo.,29
aangezien lumacaftor kan helpen bij de afgifte van Phe508del-CFTR aan het celoppervlak en ivacaftor de openingstijd en chloridegeleiding door de epitheliale cel verhoogt, kan verbetering van het onderliggende phe508del-defect mogelijk zijn met de combinatie van beide moleculen. In vitro studies van gecombineerde ivacaftor en lumacaftor in respiratoire epithelia met phe508del-mutatie hebben aangetoond dat lumacaftor het CFTR-chloridetransport alleen met 15% verhoogt, en wanneer ivacaftor wordt toegevoegd, neemt het transport toe tot bijna 30%., Deze combinatie van geneesmiddelen is onderzocht in een Fase II-studie bij patiënten met phe508del-mutatie. Volledige resultaten zijn nog niet gepubliceerd, maar initiële gegevens suggereren een gunstig effect op de longfunctie bij homozygote Phe508del, maar niet bij heterozygositeit.Er worden twee klinische onderzoeken ontwikkeld bij patiënten van 12 jaar of ouder die homozygoot zijn voor de mutatie Phe508del om gecombineerde ivacaftor en lumacaftor te evalueren; dit zijn de onderzoeken TRAFFIC (NCT01807923)31 en TRANSPORT (nct01807949)32. De resultaten kunnen ongeveer de helft van de CF populatie toelaten om CFTR modulerende therapie te krijgen., Er zijn echter studies nodig om het effect van combinatietherapie bij heterozygote individuen Phe508del te evalueren.
een ander alternatief is de corrector compound VX-661, en studies zijn momenteel aan de gang. De effectiviteit ervan wordt alleen en in combinatie met ivacaftor (VX-770) getest en de resultaten zullen binnenkort beschikbaar zijn (NCT01531673).
klasse III-Mutatiebehandelingen
Ivacaftor (VX-770) is een CFTR-potentiator die de functie van het abnormale eiwit moduleert.,Dit molecuul werd oorspronkelijk ontworpen om de CFTR-functie in culturen van respiratoire epitheelcellen te verbeteren die één enkele gly551asp-mutatie dragen.Het is het eerste geneesmiddel dat in de VS en Europa is goedgekeurd voor de behandeling van CF bij patiënten met de gly551asp mutatie. Nadat in vitro was aangetoond dat ivacaftor in staat was het chloridetransport door het celmembraan te verbeteren, werden de eerste fase II-onderzoeken uitgevoerd met 39 patiënten.In deze studie verbeterde de CFTR-eiwitfunctie drie dagen na aanvang van de behandeling, waarbij chlorideconcentraties in zweet tot normale niveaus werden bereikt., Met deze resultaten volgden twee verdere klinische onderzoeken: STRIVE-onderzoek bij 144 patiënten van 12 jaar of ouder36 en ENVISION-onderzoek met 52 kinderen tussen 6 en 11 jaar.Beide onderzoeken omvatten patiënten met ten minste één gly551asp-mutatie en FEV1 tussen 40% en 105% die aanvankelijk gedurende een periode van 14 dagen werden gevolgd en vervolgens werden gerandomiseerd naar 150 mg oraal ivacaftor of placebo tweemaal daags gedurende een periode van 48 weken., Na het voltooien van 48 weken behandeling kregen de patiënten de gelegenheid om verder te gaan in een longitudinale open-label studie, de PERSIST studie (NCT01117012), 38 gedurende 96 weken.
in STRIVE vertoonden patiënten in de ivacaftorgroep een verbetering van 10,6% in FEV1 (primair eindpunt) vanaf dag 15 van de behandeling, die werd gehandhaafd tijdens het 48 weken durende onderzoek. Verder werd een afname van de chlorideconcentratie in zweet waargenomen (gemiddeld: -48,7 mmol/L), met een verbeterde kwaliteit van leven, een 55% afname van exacerbaties en een gewichtstoename van 2,7 kg.,
de resultaten van de ENVISION-studie komen grotendeels overeen met die van de studie bij adolescenten en volwassenen, met het verschil dat de kwaliteit van leven geen statistisch verschil bereikte. De bijwerkingen die vaker werden waargenomen in de behandelingsgroep in zowel STRIVE-als ENVISION-studies waren infecties van de bovenste luchtwegen, verstopte neus, keelpijn, duizeligheid en huiduitslag., Uit de voorlopige resultaten na de eerste 12 weken van het PERSIST-onderzoek blijkt dat de verbeteringen in de longfunctie (FEV1), respiratoire symptomen en gewichtstoename bij patiënten die met ivacaftor worden behandeld, gedurende deze periode gehandhaafd blijven. Bovendien vertoonde de subgroep van patiënten die bij PERSIST overschakelden van placebo naar ivacaftor bij baseline een verbetering van 10,8% in FEV1 na 15 dagen en 13% na 12 weken, samen met een vermindering van exacerbaties.,Ondanks de goede resultaten met ivacaftor bij de behandeling van de gly551asp-mutatie bij kinderen ouder dan 6 jaar en volwassenen na 48 weken, moeten sommige problemen nog worden opgelost. Ten eerste is het medicijn niet getest bij kinderen jonger dan 6 jaar. Het lijkt echter verstandig om het defect te corrigeren voordat onherstelbare schade optreedt, gezien het feit dat de long betrokkenheid begint voor de leeftijd van zes. Een onderzoek in deze leeftijdsgroep loopt momenteel (NCT01705145).40 ten tweede, een ander alternatief zou zijn om ook andere klasse III mutaties te proberen., In dit opzicht hebben in vitro studies met negen andere mutaties zeer vergelijkbare resultaten laten zien 41, dus het is redelijk om vergelijkbare resultaten bij patiënten te verwachten. Er is een lopend fase III klinisch onderzoek bij patiënten ouder dan 6 jaar met andere klasse III mutaties (KONCONTINUE en KONNECTION studies; NCT01614470).42,43 Ten derde, hoewel er geen behandeling beschikbaar is voor de rest van de mutatieklassen (IV–VI), kunnen CFTR-potentiatoren in deze gevallen even gunstig zijn., In dit verband wordt een klinisch onderzoek uitgevoerd naar de werkzaamheid van ivacaftor in de Arg117His klasse IV mutatie (KONDUCT studie; NCT01614457).Tot slot zijn de werkzaamheid en de veiligheid op lange termijn van ivacaftor na 48 weken nog niet vastgesteld. De G551D observationele studie (GOAL; NCT01521338)45 is een observationele studie na patiënten ouder dan 6 jaar die ivacaftor kregen., Het is gericht op het rapporteren van de werkzaamheid en veiligheid van ivacaftor op lange termijn, samen met andere relevante resultaten, waaronder ontstekingsmediatoren in sputum, mucociliaire klaring en gastro-intestinale pH. resultaten worden verwacht eind 2013.
conclusies
CF is een voorbeeld van een ziekte die goed gepositioneerd is om gebruik te maken van gepersonaliseerde geneeskunde. Op dit moment zijn twee zeer verschillende benaderingen gericht op het corrigeren van het basisdefect: gentherapie gericht op het corrigeren van de genetische verandering, en therapie met moleculen gericht op het corrigeren van het functionele defect op het eiwitniveau., De laatste begint veelbelovende resultaten voor diverse molecules in ontwikkeling te tonen, en één van hen (ivacaftor) wordt reeds op de markt gebracht voor gly551asp klasse III mutatie, met uitstekende resultaten in kinderen ouder dan 6 jaar, adolescenten en volwassenen. Het uiteindelijke doel is om corrector en potentiator behandelingen te bieden voor alle CF patiënten ongeacht hun mutatie. Aangezien de resultaten van deze en andere nieuwe moleculen verschijnen, is het waarschijnlijk dat een specifieke molecule of combinatie nodig zal zijn voor elke patiënt, afhankelijk van hun bestaande mutaties., In ieder geval, de toekomst is veelbelovend, en belangrijke stappen zijn genomen in de richting van het verkrijgen van een behandeling die effectief zal werken op de oorzaak van deze ziekte.
belangenconflicten
De auteurs verklaren geen belangenconflicten.