Innledning
ferdigstillelse av det humane genom-prosjektet var et relevant milepæl for medisinsk kunnskap, å gi de opplysninger som er nødvendige for å forstå de unike egenskapene til hver enkelt.1 Den logiske konsekvensen av denne kunnskapen ville være å være i stand til å anvende spesifikke diagnostiske tester og behandling til hver enkelt pasient basert på deres individuelle genetiske informasjon. Denne nye form for medisinsk behandling kalles tilpasset medisin.,2 Men, til tross for den store utviklingen som har ført til kunnskap om den menneskelige genom, sin oversettelse til diagnostiske og personlig behandlinger har vært mindre enn forventet. I dag, skritt blir i denne retningen med to viktige tiltak: systemer biology3 og pharmacogenetics.,4 Det endelige målet med disse aktivitetene er å utvikle et medisinsk praksis som er tilpasset egenskapene til hver enkelt som kan forutsi utbruddet eller i løpet av en bestemt sykdom, tillate passende forebyggende strategier for å bli etablert, og til slutt, gjøre det mulig for pasienten å ta del i beslutningsprosesser. Dette har blitt kalt P4 medisin.5
Cystisk fibrose (CF) er fortsatt den mest vanlige og dødelige genetisk sykdom blant Kaukasiere., Det skjer med en hastighet av 1 i 2500-6000 nyfødte, avhengig av region og etnisk opprinnelse, og i en del av friske bærere, varierende mellom 1:20 og 37.6 I Spania, takket være den progressive innføring av neonatal screening programmer i ulike samfunn, en lavere forekomst av CF er nå å bli sett enn tidligere anslått i 2009, dvs., 1/4430 levendefødte i Galicia, 1/4339 i Castilla-Leon, 1/5376 i Murcia, 1/5840 i Catalonia, og 1/6602 i Balearene.7 det er anslått at Det er 70000 JF lider over hele verden.,8 sykdommen er forårsaket av mutasjoner i genet koding av regulatorisk protein cystisk fibrose transmembrane konduktans regulator (CFTR), et klorid kanal involvert i utgivelsen av adenosin trifosfat og regulering av andre ion-transport-tv. Dette proteinet er uttrykt i luftveiene epitelceller, pancreas, galleveier, svettekjertler og genitourinary system., Endring fører til en abnormitet i ion-transport, slik at pasienter produsere tykt, klebrig slim som tetter rør av det organ hvor det er plassert, og slik endring presenterer multisystemic virkninger som bestemmer bredt spekter av kliniske manifestasjoner av JF. Til tross for store fremskritt i behandlingen av CF som har ført til lengre overlevelse (nåværende median er beregnet på 37,5 år),9 det er fortsatt en lang vei å gå for å sikre at pasienter med CF har en mengde og kvalitet av livet lik som fag uten sykdom., I denne sammenheng, nye behandlinger for å redusere sykelighet og øke overlevelse er nødvendig.
CF er et eksempel på en sykdom som godt posisjonert til å dra nytte av persontilpasset medisin. På den ene siden, det er en monogenic sykdom, forårsaket av mutasjoner i et bestemt gen. Den pathophysiology av enheten er godt preget og den terapeutiske mål er klart. Videre diagnostisering av sykdommen krever genetisk testing for identifikasjon av type sykdom, så det nøyaktige genetiske defekter bestemmes i hvert enkelt tilfelle.,10
I dag, to svært forskjellige tilnærminger er rettet mot å korrigere den grunnleggende feilen: genterapi, som tar sikte på å korrigere genetisk endring, og molekylet terapi, med sikte på å korrigere funksjonelle defekter på protein-nivå. Fokus for genterapi er innføring av normale genet kopier i luftveiene av CF-pasienter. Det innebærer innsetting av et rekombinant viral vektor, DNA som har blitt trukket ut og erstattet av nye terapeutiske DNA. Viral vektor fungerer som et redskap for å sette inn den nye DNA i cellen., Ulike typer av virus, som for eksempel adenovirus eller lentivirus, har blitt brukt til dags dato. Videre, ikke-viral partikler, for eksempel nanopartikler i stand til å sette inn DNA, har også blitt utviklet.11 Men resultatene så langt har vært dårlig, fordi varigheten av uttrykk av introduserte genet var kort med begge typer vektorer.12 STORBRITANNIA Genterapi Samarbeidet er å utvikle en fase II studie for å evaluere den kliniske effekten av en optimalisert plasmidic/liposomal DNA-vektor.13 rekruttering målet er 130 pasienter og resultatene er forventet i 2014 (NCT01621867).,14
På den annen side, terapi rettet mot å gjenopprette funksjonen av CFTR proteinet har vært mer vellykket. I de senere årene, og resultatene begynner å komme gjennom på legemidler som virker direkte på CFTR proteinet. Faktisk, i januar 2012, det første stoffet til å rette Gly551Asp mutasjon feil ble markedsført i USA. I følgende avsnitt vil vi gjennomgå tilgjengelig informasjon om fremdriften av personlig medisin for CF og tilgjengelig behandlinger som tar sikte på å korrigere defekter som forårsaker sykdommen på protein-nivå., I denne gjennomgangen, nomenklaturen som brukes for beskrivelse av CFTR-genet mutasjoner som er utviklet av det Menneskelige Genom Variasjon Society15 vil bli brukt.
Mutasjoner og Protein Defekt
CF er en autosomal recessiv arvelig sykdom, slik mutasjon må være til stede i begge kopier av CFTR-genet til å bli påvirket. Til dags dato, med over 1900 CFTR-genet mutasjoner assosiert med sykdommen har blitt identifisert i den kodende sekvensen, messenger RNA eller andre elementer. Mutasjoner i CFTR-genet er tilgjengelige for konsultasjon på Cystisk Fibrose Mutasjon Database.,16 Den første mutasjonen som er beskrevet, og de mest vanlige over hele verden, er Phe508del, men det er andre spesifikke mutasjoner med varierende frekvens mellom ulike etniske grupper. I Spania, den gjennomsnittlige frekvensen av Phe508del mutasjon er mellom 50% og 60% av alle de undersøkte kromosomer, den nest hyppigste er Gly542X med 4%-8%, etterfulgt av Asn1303Lys i 2%-4% av tilfellene. Den mutasjoner er beskrevet hittil er klassifisert i seks typer eller klasser i henhold til den mekanismen som forårsaker sykdommen.17 Disse typer mutasjoner er oppsummert i Fig. 1., Klasse i-mutasjoner fører til en tidlig stopp codon i messenger-RNA som hindrer oversettelse av komplett protein. Dermed protein som produseres er korte og ikke-fungerende. Klasse II-mutasjoner kode et strukturelt unormal og misfolded protein som er fjernet av endoplasmic reticulum før nå celleoverflaten. Den vanligste mutasjon i CF, Phe508del, tilhører denne gruppen. I tilfelle av mutasjoner i klasse III til IV, proteiner nå celleoverflaten, men ikke fungerer. Klasse III mutasjoner føre til en redusert kanal aktivering, så tv forbli lukket., Klasse IV mutasjoner føre til en reduksjon i ion-konduktans gjennom kanalen. Klasse V mutasjoner kode mindre proteiner som resulterer i en redusert mengde av CFTR i celleoverflaten, slik at en bestemt funksjon skjer, men på et redusert nivå. Til slutt, klasse VI mutasjoner fører til en kortere halveringstid på grunn av protein ustabilitet, og kan også skade regulering av nærliggende CFTR-tv i celleoverflaten.
– >
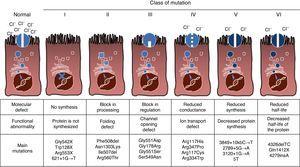
Typer mutasjoner i cystisk fibrose.
CFTR Modulatorer
Tre hovedklassene har blitt identifisert i utviklingen av legemidler for å reparere CFTR proteinet.11 Den første gruppen er for tidlig stopp codon suppressors (klasse i-mutasjoner). Disse stoffene hindrer identifikasjon av dette tidlig stopp codon, slik at proteinsyntesen kan fortsette frem til ferdigstillelse. Den andre gruppen er de CFTR korrekturlakker. Disse forbindelsene er utformet for å korrigere defekter i transport av brettet protein (klasse II-mutasjoner) til cellemembranen, der det kan bli i stand til å fungere tilnærmet normalt., Den tredje gruppen består av den såkalte CFTR potentiators. Det er disse stoffene laget for å målrette CFTR proteinet på celleoverflaten for å forbedre sin funksjon. Dermed er disse potentiators kan handle på klasse III, IV, V og VI mutasjoner. I dag, mange molekyler ved hjelp av disse ulike mekanismer som er under etterforskning, og en av disse har allerede nådd markedet: Ivacaftor (VX-770) er en CFTR potentiator godkjent i USA i januar 2012 for behandling av CF-pasienter eldre enn 6 år som har Gly551Asp mutasjon.,
Klassen jeg Mutasjon Behandlinger
Omtrent 10% av CF tilfellene er forårsaket av klasse i-mutasjoner. Den første legemidler som brukes for denne klassen var aminoglykosider. For flere år siden, gentamicin ble rapportert å ha muligheten til å maskere prematur stopp codon å hindre syntese av CFTR proteinet. Dette er oppnådd ved innsetting av en aminosyre som gjør at ribosomet til å fortsette å lese genet, og produserer en full-lengde protein. Prekliniske studier har vist at proteinet kan bli syntetisert i en mus modell og 35% av protein funksjon ble gjenopprettet in vitro.,18,19 effekten av intravenøs administrasjon av gentamicin ble vurdert i to studier av CF-pasienter med ulike typer av klasse i-mutasjoner. Den ene ble gjennomført i USA i fem pasienter,20 og en annen, inkludert 18 pasienter i Frankrike.21 Men, selv om responsen var positiv, resultatene varierte mye, slik at fordelen var ikke universelle. Videre, toksisitet problemer med aminoglykosider bidratt til en ugunstig profil.
Et syntetisk alternativ er ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., Dette er et molekyl som er designet for å aktivere ribosomes å lese den genetiske informasjon mens du «hoppe over» den prematur stopp codon, produsere en funksjonell CFTR proteinet.22 farmakokinetikk ataluren har blitt vist i dyremodeller og i fase II studier. I to små foreløpige studier,23,24 en gruppe av CF-pasienter som behandles med orale ataluren viste forbedring av electrophysiological misdannelser av sykdom, med en økning i antall celler i nesen å uttrykke protein på overflaten deres., Senere, en annen liten studie av 19 pasienter vurdert ulike doser av ataluren oralt hver 8h, med forbedringer i CFTR aktivitet og kliniske parametre og en god sikkerhetsprofil.25 resultater av en fase III klinisk studie av ataluren som ennå ikke hadde blitt formelt publisert ble rapportert i 2012 North American Cystisk Fibrose Konferanse. Av en total av 238 pasienter eldre enn 6 år ble randomisert til å motta ataluren 10, 10, 20 mg/kg eller placebo hver 8h i 48 uker., Det var ingen signifikante forskjeller i FEV1 i ataluren versus placebo etter 48 ukers behandling (-2.5% ataluren vs -5.5% for placebo, P=ns). Når stratifisert med kronisk nebulized bruk av antibiotika, det var en 6.7% forskjell i gjennomsnittlig endring etter 48 uker i favør av ataluren for pasienter som ikke på tobramycin, mens det var ingen forskjell i endring i FEV1 i de mottar nebulized tobramycin. Noe liknende skjedde med andelen av eksaserbasjoner, hvor forskjellen mellom begge gruppene var betydelig., Når stratifisert, pasientene i ataluren-gruppe som ikke finnes på nebulized tobramycin viste en prosentvis nedgang i eksaserbasjoner på 43% sammenlignet med placebo-gruppen. Nese potensiell forskjell og svette-klorid tester viste ingen forskjell mellom gruppene, uavhengig av bruken av nebulized antibiotika. Forfatterne konkluderte med at fordelene var større hos pasienter som ikke får kronisk behandling med antibiotika med nebulized aminoglycoside, spekulerer at tobramycin og ataluren kommuniserte på et ribosom-nivå, produsere antagonisme når de brukes samtidig.,26
Klasse II Mutasjon Behandlinger
Klasse II mutasjoner, som er den mest hyppige mutasjon av sykdom (Phe508del), er til stede i et stort nummer av CF-pasienter, noe som gjør dem et primært mål i CF forskning. Ulike molekyler som har vært studert, og de fleste som har blitt utviklet av Vertex Pharmaceuticals Inc. Den første corrector sammensatte, lumacaftor (VX-809), viste god in vitro effekt, bedre klorid transport i 14%.,27 Men resultatene var litt skuffende i pasienter, fordi forbedringer i klorid konsentrasjon i svette var veldig liten (7mmol/l), og det var ingen endringer i nese potensial.28
effekter av CFTR potentiator ivacaftor (VX-770), et stoff som for klasse III mutasjoner (se beskrivelse nedenfor), har også vært undersøkt hos pasienter som var homozygote for Phe508del. Resultatene av DISCOVER studie viser at ivacaftor er ikke forbundet med en betydelig forbedring i FEV1, livskvalitet eller antall av eksaserbasjoner versus placebo.,29
Siden lumacaftor kan hjelpe levering av Phe508del-CFTR til celleoverflaten og ivacaftor øker åpning tid og klorid ledning gjennom epiteliale celle, forbedring av underliggende Phe508del feil kan det være mulig med kombinasjon av begge molekyler. In vitro-studier av kombinert ivacaftor og lumacaftor i luftveiene epithelia med Phe508del mutasjon har vist at lumacaftor øker CFTR klorid transport med 15% på sin egen, og når ivacaftor er lagt til, transport øker til nesten 30%., Denne kombinasjonen av stoffene har vært undersøkt i en fase II-studie på pasienter med Phe508del mutasjon. Fullstendige resultater har ikke blitt publisert ennå, men foreløpige data tyder på en gunstig effekt på lungefunksjon i homozygote Phe508del, men ikke i heterozygosity.30 To kliniske studier hos pasienter som er 12 år eller eldre homozygote for mutasjonen Phe508del er utviklet for å evaluere kombinert ivacaftor og lumacaftor; disse er TRAFIKKEN (NCT01807923)31 og TRANSPORT (NCT01807949)32 studier. Resultatene kan føre til at om lag halvparten av CF-befolkningen til å motta CFTR modulerende behandling., Men studier er nødvendig for å evaluere effekten av kombinasjonsbehandling i Phe508del heterozygote individer.
et Annet alternativ er corrector sammensatte VX-661, og studiene er for tiden pågår. Virkningen er blitt testet ut alene og i kombinasjon med ivacaftor (VX-770), og resultatene vil snart være tilgjengelig (NCT01531673).33
Klasse III Mutasjon Behandlinger
Ivacaftor (VX-770) er en CFTR potentiator som modulerer funksjon av unormal protein.,34 Dette molekylet var opprinnelig designet for å forbedre CFTR-funksjonen i kulturer av respiratorisk epitel celler som bærer en enkelt Gly551Asp mutasjon.34 Det er det første stoffet godkjent i USA og Europa for behandling av CF-pasienter bærer Gly551Asp mutasjon. Etter kapasiteten på ivacaftor å forbedre klorid transport gjennom cellemembranen ble vist in vitro, første fase II studier ble gjennomført med 39 pasienter.35 I denne studien, CFTR proteinet funksjon forbedret tre dager etter påbegynt behandling, nådde klorid konsentrasjoner i svette av opp til normalt nivå., Med disse resultatene ytterligere to kliniske studier som følges: STREBER etter studiet, i 144 pasienter i alderen 12 år eller more36 og SER for studiet, inkludert 52 barn mellom 6 og 11 år.37 Begge studiene inkluderte pasienter med minst én Gly551Asp mutasjon og FEV1 mellom 40% og 105% som opprinnelig ble fulgt over en periode på 14 dager og deretter randomisert til å motta 150mg av oral ivacaftor eller placebo to ganger om dagen i en periode på 48 uker., Etter å ha fullført 48 ukers behandling, og pasienter ble gitt mulighet til å fortsette i en langsgående åpne-label studien, VEDVARER studie (NCT01117012),38 96 uker.
I STREBE etter, pasienter i ivacaftor gruppen hadde en økning på 10,6% i FEV1 (primære endepunkt) starter fra dag 15 av behandling, som ble opprettholdt i løpet av 48-ukers studie. Videre, en nedgang i sulfat konsentrasjonen i svette ble observert (betyr: -48.7 mmol/L), med forbedret kvalitet av livet, en 55% reduksjon i eksaserbasjoner og en vektøkning på 2,7 kg.,
resultatene av studien SER sammenfaller i stor grad med de av studiet i ungdom og voksne, med den forskjellen at kvaliteten på livet nådde ikke statistisk forskjell. Det ugunstig virkninger oftere observert i behandlingen gruppen i begge STREVER og SER for studier ble øvre luftveisinfeksjoner, tett nese, sår hals, svimmelhet og hudutslett., Den foreløpige resultater etter de første 12 uker av den VEDVARER studie viser at forbedringer i lungefunksjon (FEV1), respirasjonssymptomer, og vektøkning hos pasienter behandlet med ivacaftor er opprettholdt i denne perioden. Videre undergruppe av pasienter som byttet fra placebo for å ivacaftor ved baseline i VEDVARE opplevd en 10.8% forbedring i FEV1 på 15 dager, og 13% ved 12 uker, sammen med en reduksjon i eksaserbasjoner.,39
til Tross for de gode resultatene med ivacaftor i behandling av Gly551Asp mutasjon hos barn eldre enn 6 år og voksne i 48 uker, noen spørsmål gjenstår å løses. For det første, at stoffet har ikke blitt testet på barn under 6 år. Men, det virker fornuftig å rette opp feilen før irreversible skader oppstår, vurderer at lunge engasjement starter før fylte seks. En studie i denne aldersgruppen er for tiden pågår (NCT01705145).40 for det Andre, et annet alternativ ville være å også prøve andre klasse III mutasjoner., I denne forbindelse, in vitro studier på ni andre mutasjoner har vist svært lik resultatene,41, så det er rimelig å forvente tilsvarende resultater hos pasienter. Det er en pågående fase III klinisk studie hos pasienter eldre enn 6 år, med andre klasse III mutasjoner (KONTINUE og KONNECTION studier; NCT01614470).42,43 for det Tredje, selv om ingen behandling er tilgjengelig for resten av mutasjon klasser (IV–VI), CFTR potentiators kan være like gunstig i disse tilfellene., En klinisk studie er gjennomført i forbindelse med dette, evaluere effekten av ivacaftor i Arg117His klasse IV mutasjon (KONDUCT studien, NCT01614457).44 til Slutt, effektivitet og langsiktig sikkerhet for ivacaftor utover 48 uker har ennå ikke blitt etablert. Den G551D Observasjonsstudie (MÅL; NCT01521338)45 er en observasjonsstudie følgende pasienter eldre enn 6 år mottar ivacaftor., Det er rettet mot rapportering av effekt og sikkerhet av langsiktig ivacaftor, sammen med andre utfall av interesse som inkluderer inflammatoriske mediatorer i sputum, mucociliary klaring, og fordøyelsessystemet pH. Resultatene er forventet i slutten av 2013.
Konklusjon
CF er et eksempel på en sykdom som godt posisjonert til å dra nytte av persontilpasset medisin. I dag, to svært ulike tilnærminger som tar sikte på å korrigere den grunnleggende feilen: gen terapi som tar sikte på å korrigere genetisk endring, og behandling med molekyler som tar sikte på å korrigere funksjonelle defekter på protein-nivå., Sistnevnte er i ferd med å vise lovende resultater for ulike molekyler i utvikling, og en av dem (ivacaftor) er allerede markedsført for Gly551Asp klasse III mutasjon, med gode resultater hos barn eldre enn 6 år, ungdom og voksne. Det endelige målet er å gi corrector og potentiator behandlinger for alle CF-pasienter, uansett deres mutasjon. Som resultatene av disse og andre nye molekyler vises, er det sannsynlig at en bestemt molekyl eller en kombinasjon vil være nødvendig for hver pasient, avhengig av deres eksisterende mutasjoner., I alle fall, fremtiden er lovende, og viktig skritt er tatt mot å få en behandling som effektivt handle på grunn av denne sykdommen.
Interessekonflikter
forfatterne erklærer ingen interessekonflikter.