Introduzione
Nuclear factor kappa B (NF-kB) è un antico fattore di trascrizione proteica (Salminen et al., 2008) e considerato un regolatore dell’immunità innata (Baltimora, 2009). La via di segnalazione NF-kB collega i segnali patogeni e i segnali di pericolo cellulare organizzando così la resistenza cellulare agli agenti patogeni invasori., In effetti, una pletora di studi ha dimostrato che NF-kB è un hub di rete responsabile della complessa segnalazione biologica (Albensi e Mattson, 2000; Kaltschmidt e Kaltschmidt, 2009; Karin, 2009). A tal fine, è stato ipotizzato che NF-kB sia un regolatore principale delle cascate biochimiche conservate evolutivamente (Mattson et al., 2000). Altri fattori sono anche traslocati nei mitocondri e sono coinvolti nella modulazione dell’espressione (Barshad et al., 2018a), ma non sono al centro di questa recensione., Lo scopo di questa recensione è quello di tentare di capire come l’attività di NF-kB contribuisce alla funzione mitocondriale. Si presume che il lettore abbia già una comprensione della biologia mitocondriale di base. Nel caso di ulteriori studi, il lettore si riferisce a molti studi e recensioni eccellenti sulla struttura e la funzione mitocondriale (Hall, 1979; Fox, 1982; Roger e Silberman, 2002; Henze e Martin, 2003; Conradd, 2006; Ettema, 2016; Wang e Youle, 2016; Barshad et al., 2018b).,
Attivazione NF-kB
Fattore nucleare kappa B subunità, comprendente il complesso NF-kB, sono espressi in entrambi i neuroni e glia. Il complesso NF-kB esiste in uno stato inattivo nel citoplasma (Ghosh et al., 1998; Aggarwal et al., 2004; Hayden e Ghosh, 2004) dove l’attivazione di NF-kB è stata ben descritta (Li e Karin, 2000; Baud e Jacque, 2008; Israele, 2010). Quando stimolato da molecole come il TNFa o altri fattori di stress cellulare, il TNFa si lega ai recettori del TNF (Figura 1)., Questo legame, attraverso diversi passaggi intermedi, porta ad un’interazione con il complesso IKB chinasi (IKK), che poi porta alla fosforilazione di IkB, e successivamente si traduce in IKB ubiquitinazione e degradazione. Una volta degradato, il dimero NF-kB rimanente (ad esempio, subunità p65 / p50) si trasferisce al nucleo, dove si lega alla sequenza di consenso del DNA di vari geni bersaglio. La selettività della risposta NF-kB si basa su diversi fattori (Sen e Smale, 2010) tra cui la composizione del dimero, i tempi e il tipo di cella., L’influenza di NF-kB sulla sopravvivenza cellulare è anche complessa e può essere neuroprotettiva o proinfiammatoria, a seconda del tipo di cellula, dello stadio di sviluppo e dello stato patologico (Qin et al., 2007).
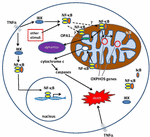
Figura 1. Vie per la segnalazione del fattore nucleare κ B (NF-kB) nel citoplasma e nel mitocondrio. Il complesso tri-subunt NF-kB (ad esempio, p65, p50, IkB – una possibile combinazione) esiste in uno stato inattivo nel citoplasma. L’attivazione di NF-kB viene avviata quando molecole come il TNFa si legano ai recettori del TNF (esistono diversi tipi)., Altri stimoli esterni o interni possono anche attivare NF-kB. Un complicato processo di trasduzione del segnale inizia quindi una volta attivati i recettori TNF; la chinasi IkB (IKK) viene infine attivata e porta alla fosforilazione di IkB, che si traduce in ubiquitinazione e degradazione di IkB. Una volta che IkB è degradato, il dimero NF-kB rimanente (ad esempio, p65/p50 o p50/p50 sono possibili combinazioni di subunità) si trasferisce al nucleo, dove si lega a una sequenza di consenso del DNA di geni bersaglio., Per processi non ben compresi, il complesso NF-kB o le subunità NF-kB possono anche migrare nel mitocondrio, dove l’evidenza suggerisce che occupa lo spazio intermembrana. Una volta all’interno dei mitocondri, si pensa che NF-kB interagisca con i geni OXPHOS (mtDNA mitocondriale) che porta all’espressione di proteine coinvolte in varie funzioni, tra cui la dinamica mitocondriale e la regolazione della COX III (componente del Complesso IV)., L’evidenza suggerisce anche che NF-kB può funzionare come un interruttore nei mitocondri e controllare l’equilibrio tra l’utilizzo della glicolisi citoplasmatica e la respirazione mitocondriale nelle cellule normali e nel cancro. Infine, i dati indicano anche la stimolazione intrinseca della via apoptotica, dove l’attivazione di NF-kB nei mitocondri porta al rilascio del citocromo c, innescando così le cascate di caspasi e la morte cellulare programmata.,
dal punto di vista Organizzativo, NF-kB è un Rel fattore di trascrizione della famiglia ed è associato con cinque geni, NF-kB1, NF-kB2, REAL, RELB, e REL (Chen e Greene, 2004); questi geni codificano per diverse proteine, NF-kB1, NF-kB2, Real, RelB, e c-Rel, rispettivamente, dove due di queste proteine sono grandi precursori delle proteine denominate p105 e p100 che subiscono proteolisi per diventare p50 e p52, rispettivamente. Queste proteine contengono domini di REL-omologia (RHD) nella loro regione amino-terminale; la regione RHD è composta da 2 domini separati, ma adiacenti., La sequenza più distante dalla regione carbossi-terminale consente alla proteina di legarsi al DNA. Una sequenza più interna consente alle proteine della famiglia Rel di dimerizzare (omo-o eterodimeri) per la soppressione dell’espressione tramite il legame della loro corrispondente famiglia di inibitori, le proteine IkB (Chen e Greene, 2004). Quest’ultima sequenza include la sequenza di localizzazione nucleare (NLS) che viene smascherata quando IkB non è legato dalla degradazione. L’NLS ha il compito di guidare o etichettare le proteine attive per l’importazione nel nucleo cellulare (Chen e Greene, 2004; Karin et al., 2004; Barger et al.,, 2005).
Tre di queste proteine (RelA, RelB e c-REL) codificano anche un dominio di transattivazione (TADs) nella loro regione carbossi-terminale. I TAD consentono a queste proteine di interagire con l’apparato di trascrizione basale, noto come TATA binding protein (TBP), Fattore di trascrizione IIB, nonché i co-attivatori trascrizionali p300 e cAMP response element (CREB) binding protein (CBP) (Chen e Greene, 2004)., Solo queste tre proteine sono in grado di indurre la trascrizione delle loro regioni codificanti il DNA mentre le altre proteine, gli omodimeri p50 e p52, sono in grado di occupare i siti di legame del DNA senza avviare la trascrizione. Detto questo, le successive 2 proteine omodimeri di p50 e p52 agiscono come repressori trascrizionali (Chen e Greene, 2004).
Gli omodimeri p105 e p100 occupano siti di legame del DNA bloccando così la trascrizione tramite fattori di trascrizione che possiedono TADs (Barger et al., 2005). Una terza forma di repressione trascrizionale è dovuta alle proteine IkB., Queste proteine hanno diverse ripetizioni di ankyrin come dominio principale e funzione legandosi al RHD che maschera l’NLS (Karin et al., 2004). Senza un NLS attivo, le proteine NF-kB sono limitate al citoplasma e non sono in grado di migrare nel nucleo e quindi la trascrizione è bloccata.
NF-kB si trova nei mitocondri
Nel 2001, uno studio di Bottero et al. (2001) ha trovato IkBa e la subunità NF-kB p65 in frazioni subcellulari e mitocondri purificati dalle cellule di Jurkat. Le cellule di Jurkat sono una linea cellulare immortalata di cellule linfocitarie T umane che vengono utilizzate per studiare la leucemia., Nello studio di Bottero, è stato determinato che IkBa e NF-kB p65 erano localizzati nello spazio intermembrana mitocondriale. Lo spazio intermembrana mitocondriale è lo spazio che esiste tra la membrana mitocondriale interna (IMM) e la membrana mitocondriale esterna (OMM).
Successivamente, Cogswell et al. (2003) ha anche mostrato che le subunità NF-kB, p50 e p65 e IkBa, sono state trovate nei mitocondri. Per determinare ciò, sono stati utilizzati diversi metodi per fornire prove, inclusa la microscopia elettronica di sezioni di cellule U937., Le cellule U937 sono state isolate per la prima volta dal linfoma di un paziente maschio di mezza età per studiare il comportamento e la differenziazione dei monociti. Qui Cogswell et al. (2003) è stato in grado di visualizzare le subunità NF-kB p50 e p65 e IkBa nella matrice interna dei mitocondri. In questo studio sono state esaminate anche cellule epatiche di ratto e sono state identificate anche la subunità p50 e la subunità IkBa. Inoltre, le cellule U937 sono state stimolate per 1 h con TNFa, un trigger noto della via di segnalazione NF-kB., In questo esperimento, le analisi Western blot nelle frazioni mitocondriali e citoplasmatiche hanno rilevato che il trattamento con TNFa ha causato una perdita di IkBa in entrambi i compartimenti mitocondriali e citoplasmatici di 30 min dopo il trattamento, suggerendo che IkBa era degradato. L’analisi EMSA, un test in vitro che rileva l’attivazione di NF-kB e il legame non specifico alle sequenze di DNA, è stata condotta anche su proteine prelevate da estratti nucleari da mitocondri isolati da cellule U937 stimolate con TNFa., Qui hanno determinato la segnalazione TNFa ha portato ad un aumento dell’attività di legame del DNA di NF-kB p50, in proteine prelevate dai mitocondri.
Altri studi hanno anche rilevato NF-kB nei mitocondri. Questi includono studi (Guseva et al., 2004; Zamora et al., 2004) nelle linee cellulari umane del fibroblasto HT1080, nelle linee cellulari umane della prostata LNCAP e PC3 e nelle cellule di HeLa. Nelle cellule LNCaP sono state trovate subunità mitocondriali NF-kB p50 e p65 legate al DNA mitocondriale (mtDNA)., Presi insieme, questi studi mostrano la prova per la segnalazione di NF-kB nei mitocondri e che NF-kB regola l’espressione mitocondriale dell’mRNA (vedi NF-kB e la sezione di espressione genica mitocondriale sotto).
NF-kB Controlla la dinamica mitocondriale
Ci sono diverse proteine coinvolte nella dinamica (fissione e fusione) e nella morfologia dei mitocondri (Karbowski e Youle, 2003; Olichon et al., 2006; Brooks e Dong, 2007; Song et al., 2008; Autret e Martin, 2010; Silva et al., 2013; Sinha e Manoj, 2019). Uno di questi è la proteina atrofia ottica 1 (OPA1) (Olichon et al.,, 2006; Garcia et al., 2018; Lee e Yoon, 2018). Gli studi hanno suggerito che OPA1 è un regolatore della fusione della membrana interna mitocondriale e anche del rimodellamento delle cristae mitocondriali (Cipolat et al., 2006). Recentemente Laforge et al. (2016) ha dimostrato che l’assenza di IKKa ha avuto un impatto sull’espressione di OPA1 nei mitocondri e sulla morfologia mitocondriale.
Sorprendentemente, in un recente studio di Nan et al. (2017), la stimolazione del recettore TNFa 2 (TNFR2) è stata trovata per promuovere la fusione mitocondriale tramite l’attivazione NF-kB-dipendente dell’espressione OPA1 nei miociti cardiaci., È importante sottolineare che l’attivazione del TNFR2 in questo studio ha protetto i miociti cardiaci dallo stress regolando l’espressione OPA1. Somministrando basse concentrazioni di TNFa esogeno (0,5 ng / mL) prima dell’ischemia-riperfusione sembrava migliorare la sopravvivenza cellulare, mentre concentrazioni più elevate (10-20 ng/mL) hanno portato ad effetti tossici nelle cellule.
NF-kB e Apoptosi nei mitocondri
Il ruolo dei mitocondri nella morte cellulare programmata, o apoptosi, è noto da molto tempo (Green e Reed, 1998; Wang e Chen, 2015)., Il ruolo più importante per i mitocondri è la generazione di ATP; tuttavia, la seconda funzione più importante per i mitocondri è probabilmente nel controllo della morte cellulare. Come fa il mitocondrio a farlo? Se il mitocondrio non riesce a innescare la morte cellulare, il cancro è spesso la conseguenza. Quindi, al fine di regolare la morte cellulare, i mitocondri integrano segnali provenienti da una varietà di fonti, che sono noti come vie intrinseche di apoptosi. I componenti dell’attività di NF-kB sembrano essere uno di questi segnali, sebbene il TNFa, un attivatore di NF-kB, faccia parte di una via estrinseca di apoptosi., Le vie estrinseche (mediate dal recettore della morte) sono iniziate all’esterno della cellula, mentre le vie intrinseche dell’apoptosi sono mediate e innescate nei mitocondri.
In un recente studio di Pazarentzos et al. (2014), IkBa è stato trovato per esercitare attività pro-apoptotica in quanto ha inibito l’anti-apoptotico NF-kB. Nella maggior parte delle cellule, l’attivazione di NF-kB porta all’espressione genica target a valle che innesca la resistenza alla morte cellulare (Luo et al., 2005). In questo studio, è stato dimostrato che una nuova funzione di apoptosi era dovuta a IkBa, la subunità che inibisce l’attivazione di NF-kB. Pazarentzos et al., (2014) ha scoperto che IkBa si localizza all’OMM dove interagisce con un canale anionico dipendente dalla tensione (VDAC) e l’esochinasi II mitocondriale (HKII) per stabilizzare questo complesso e prevenire il rilascio del citocromo c mediato da Bax per l’apoptosi. Bax è un membro della famiglia di proteine Bcl-2, che hanno dimostrato di essere regolatori della morte cellulare programmata (Karbowski et al., 2006).
Altri studi hanno anche accennato al ruolo di NF-kB nella regolazione più diretta dell’apoptosi nel mitocondrio. In uno studio di Liu et al., (2004), l’inibizione di NF-kB da sola nei macrofagi ha portato al rilascio del citocromo c. Ricordiamo che il citocromo c è responsabile della spola di elettroni dal Complesso III al complesso IV e che il rilascio del citocromo c nel citoplasma, un attivatore delle caspasi, è un passo chiave nell’innescare l’apoptosi.
NF-kB e respirazione mitocondriale
Il fattore nucleare kappa B ha dimostrato in molti studi di promuovere la tumorigenesi. Come questo accade non era esattamente chiaro. In uno studio innovativo di Mauro et al., (2011), NF-kB è stato trovato per upregulate la respirazione mitocondriale nelle cellule di carcinoma del colon. Qui hanno stabilito che questa funzione di NF-kB sopprime l’effetto Warburg. Ricordiamo che l’effetto Warburg (Vander Heiden et al., 2009) descrive l’osservazione che le cellule tumorali tendono a favorire il metabolismo per glicolisi piuttosto che per la via di fosforilazione ossidativa più efficiente. Quindi in questo studio gli autori hanno determinato che NF-kB organizza reti di metabolismo energetico controllando l’equilibrio tra l’utilizzo della glicolisi e la respirazione mitocondriale., È interessante notare che hanno trovato un ruolo per NF-kB nell’adattamento metabolico nelle cellule normali e nel cancro. I loro risultati hanno inoltre suggerito che la soppressione del metabolismo mitocondriale nelle cellule tumorali consolidate mediante inibizione di NF-kB e metformina diminuisce la tumorigenesi.
NF-kB e espressione genica mitocondriale
Il fattore nucleare kappa B è un noto regolatore dell’espressione genica – sia negativamente che positivamente (Mattson et al., 2000). Tuttavia, il modo in cui NF-kB regola o influenza l’espressione genica mitocondriale codificata nucleare è meno compreso., mtDNA umano possiede 37 geni che codificano per 13 polipeptidi. È stato dimostrato che i geni mtDNA codificano per molte delle subunità di tutti e 5 i complessi della catena di trasporto degli elettroni (ETC), 2 rRNA e 22 TRNA. Anche se, la maggior parte delle subunità ETC sono codificate dal DNA nucleare, che potrebbe essere influenzato dall’attività NF-kB (Calvo et al., 2016).
Ad esempio, è stato affermato (Cogswell et al., 2003) che la via NF-kB può regolare negativamente l’espressione genica mitocondriale associata alla subunità COX III., La subunità di COX III è codificata da mtDNA ed è un componente del complesso IV nel mitocondriale ECC. Funziona come una subunità catalitica nel complesso IV, che è il complesso associato al consumo di ossigeno mitocondriale. In uno studio di Cogswell et al. (2003), la modulazione dell’attivazione di NF-kB ha provocato la perdita di espressione sia di COX III che di mRNA del citocromo B. Altri studi supportano un ruolo per NF-kB che regola geni mitocondriali aggiuntivi, come COX I e Cytb (Psarra e Sekeris, 2008, 2009; Barshad et al., 2018b)., Inoltre, la subunità NF-kB p65 ha ridotto i livelli di mRNA CytB codificato con mtDNA, possibilmente legandosi al D-loop nelle cellule umane in assenza di p53 (Johnson et al., 2011). Nel complesso, questi risultati suggeriscono che la segnalazione NF-kB può influenzare l’attività enzimatica dei complessi ETC respiratori.
NF-kB Media la disfunzione indotta da Aß nei mitocondri
La malattia di Alzheimer (AD) è associata all’accumulo di placche Aß e / o alla comparsa di grovigli neurofibrillari (NFTs) in alcune regioni del cervello (Duyckaerts et al., 2009)., Tuttavia, esiste una controversia sul fatto che Aß sia un agente causale dell’AD o se Aß sia semplicemente correlato all’invecchiamento. Accumulare prove (Aliev et al., 2009; Correia et al., 2012; Cadonic et al., 2016; Cardoso et al., 2017; Djordjevic et al., 2017) ora punta anche a cambiamenti nel metabolismo cerebrale guidati dalla disfunzione mitocondriale come un processo centrale per molti disturbi neurodegenerativi legati all’età, tra cui l’AD. Aggiungendo a questa evidenza, ci sono anche menomazioni nell’attività enzimatica dei complessi proteici dell’ETC e alterazioni nell’attività enzimatica antiossidante (Kolosova et al.,, 2017) in AD. In particolare, l’attività IV complessa ha dimostrato di essere influenzata negativamente nell’AD (Mutisya et al., 1994).
In un recente studio di Shi et al. (2014), è stato riscontrato che Aß comprometteva la funzione mitocondriale tramite segnalazione NF-kB. Inoltre, Shi et al. (2014) ha mostrato qui che Aß ha diminuito l’espressione della subunità COX III tramite un percorso NF-kB., È importante sottolineare che, per eliminare la possibilità che IkBa sia stata fosforilata da Aß nel citoplasma (e quindi trasportata nei mitocondri), i mitocondri isolati sono stati incubati con Aß in presenza (o assenza) di un bloccante NF-kB, vale a dire BAY11-7082. Qui hanno trovato la fosforilazione indotta da Aß e la degradazione di IkBa nei mitocondri isolati.
Questi risultati hanno anche importanti implicazioni per il trattamento dell’AD, come dimostrato da recenti studi di Snow et al. (2018) e come mostrato in altri studi correlati (Djordjevic et al., 2017; Adlimoghaddam et al.,, 2019) che suggeriscono il targeting della segnalazione NF-kB nei mitocondri può avere un valore terapeutico. Ad esempio, in Snow et al.lo studio, creatina-un noto modulatore della funzione mitocondriale (Tarnopolsky e Beal, 2001), ha dimostrato di aumentare e alterare positivamente i livelli proteici di CAMKII, PSD-95 e subunità 1 complesse nei topi alimentati con creatina, mentre la subunità IkB inibitoria NF-kB era diminuita. Per ulteriori letture sul potenziale effetto terapeutico della creatina sulla funzione mitocondriale e nei disturbi mitocondriali o altri disturbi neurologici vedere studi e recensioni di Matthews et al., (1998), Klivenyi et al. (1999), Tarnopolsky e Beal (2001), Hersch et al. (2006), Rodriguez et al. (2007) e Beal (2011).
Il ruolo di NF-kB nell’infiammazione e nel metabolismo mitocondriale
Dati crescenti (Lamas et al., 2003; Mauro et al., 2011; Moretti et al., 2012) suggeriscono che la segnalazione NF-kB, che è un mediatore dei processi infiammatori, funziona anche come regolatore e integratore con il metabolismo energetico. In un recente studio di Zhong et al. (2016), NF-kB ha dimostrato di limitare l’attivazione dell’infiammasoma attraverso l’eliminazione dei mitocondri danneggiati., Sorprendentemente, NF-kB è apparso sia per innescare il NLRP-3 inflammasome per l’attivazione e anche impedito eccessiva infiammazione e trattenuto NLRP-3 inflammasome attivazione; anche se il meccanismo di ritenuta è stato mal definito. Qui è stato ipotizzato che oltre a NF-kB essendo un attivatore di geni infiammatori, ha anche funzionato in questo studio limitando l’attivazione dell’infiammasoma NLRP3 e la produzione di IL-1β. Inoltre, è stato riscontrato che l’induzione p62 era responsabile dell’attività inibitoria dell’infiammasoma da parte di NF-kB., Sembra che NF-kB possa frenare la propria infiammazione nei macrofagi promuovendo la rimozione mediata da p62 dei mitocondri danneggiati (mitofagia) dopo che i macrofagi interagiscono con diversi attivatori dell’infiammasoma NLRP3.
Conclusione
Oltre 10 anni fa, NF-kB è stato rilevato nei mitocondri. Sorprendentemente, per un fattore di trascrizione così importante, sono stati fatti pochi progressi nello scoprire ruoli specifici per NF-kB che interessano il mitocondrio., Alcuni studi, come descritto sopra, forniscono prove per NF-kB nella dinamica mitocondriale, nell’apoptosi, nel controllo respiratorio, nell’espressione genica e nei meccanismi della malattia (Figura 1). Tuttavia, la duplicazione di questi risultati e la convalida generale sono ancora necessarie da parte di altri laboratori. Alcune informazioni aggiuntive possono essere raccolte dal fatto che altri fattori di trascrizione che hanno effetti sui geni nucleari, come AP-1, p53, CREB, c-Myc, Wnt13, Dok-4, HMGA1 e c-Src sono stati rilevati anche nei mitocondri (Psarra e Sekeris, 2008)., È interessante notare che sono stati determinati siti di legame nel genoma mitocondriale (omologhi ai loro siti di legame nel DNA nucleare) per alcuni di questi fattori (Psarra e Sekeris, 2008) in cui si sospettano ruoli per la trascrizione mitocondriale e l’apoptosi e mostrano alcuni modelli generali di attività. Ad esempio, si può argomentare che alcuni di questi fattori (NF-kB, CREB e AP-1) si legano ai genomi mitocondriali e per lo più attenuano l’espressione genica mitocondriale (Blumberg et al., 2014), pur avendo effetti stimolanti sulla trascrizione del gene nucleare., Tuttavia, è chiaramente necessario più lavoro non solo per trovare ruoli precisi di attività, ma anche per determinare se esistono veramente modelli generali di attività.
Dopo aver esaminato questa letteratura, diventa anche evidente che il ruolo di NF-kB nella regolazione della respirazione mitocondriale ha profonde implicazioni e dimostra un livello di complessità non precedentemente apprezzato. Per esempio, Mauro et al. (2011) i dati stabiliscono un ruolo per NF-kB nell’adattamento metabolico nelle cellule normali e nel cancro e suggeriscono anche conseguenze per altri stati patologici come AD., Inoltre, dato che NF-kB può frenare la propria infiammazione come mostrato da Zhong et al. (2016), non solo è sorprendente, ma esemplifica ulteriormente la complessità della segnalazione NF-kB nella funzione mitocondriale.
In questa recensione, sono stati esaminati studi sul ruolo di NF-kB nella funzione mitocondriale e sembra che la ricerca in questo settore sia in aumento. A complicare i risultati è però l’osservazione che più fattori stanno giocando ruoli simili nella funzione mitocondriale e quindi sono necessari studi dettagliati specifici per ciascun fattore., In conclusione, possiamo porre nuovamente la domanda: cosa sta facendo NF-kB nel mitocondrio? La risposta immediata e abbreviata sarebbe-molto!
Contributi dell’autore
L’autore ha creato l’argomento e ha scritto il manoscritto.
Finanziamento
Questo lavoro è stato sostenuto dai finanziamenti del Canadian Institutes of Health Research (CIHR), Canadian Agricultural Partnership (CAP), St. Boniface Hospital Research Foundation, Alzheimer Society of Manitoba, Research Manitoba, The Honourable Douglas e Patricia Everett, e Royal Canadian Properties Limited Endowment Fund.,
Dichiarazione sul conflitto di interessi
L’autore dichiara che la ricerca è stata condotta in assenza di rapporti commerciali o finanziari che potrebbero essere interpretati come un potenziale conflitto di interessi.
Ringraziamenti
L’autore desidera ringraziare il Dr. Grant Hatch per una revisione del manoscritto.
Hayden, MS, e Ghosh, S. (2004). Segnalazione a NF-kappaB. Geni Dev. 18, 2195–2224.
PubMed Abstract/Google Scholar
Wang, X. e Chen, XJ (2015)., Una rete citosolica che sopprime lo stress proteostatico mediato dai mitocondri e la morte cellulare. Natura 524, 481-484. doi: 10.1038/nature14859
PubMed Abstract / CrossRef Full Text / Google Scholar