Einführung
Nuclear factor kappa B (NF-kB) ist eine Antike protein transcription factor (Salminen et al. 2008) und als regulator der angeborenen Immunität (Baltimore, 2009). Der NF-kB-Signalweg verbindet pathogene Signale und zelluläre Gefahrensignale und organisiert so die zelluläre Resistenz gegen eindringende Krankheitserreger., Tatsächlich hat eine Vielzahl von Studien gezeigt, dass NF-kB ein Netzwerkknotenpunkt ist, der für komplexe biologische Signalisierung verantwortlich ist (Albensi und Mattson, 2000; Kaltschmidt und Kaltschmidt, 2009; Karin, 2009). Zu diesem Zweck wurde angenommen, dass NF-kB ein Masterregulator evolutionär konservierter biochemischer Kaskaden ist (Mattson et al., 2000). Andere Faktoren werden ebenfalls in die Mitochondrien transloziert und sind an der Modulation der Expression beteiligt (Barshad et al., 2018a), stehen aber nicht im Mittelpunkt dieser Überprüfung., Der Zweck dieser Überprüfung ist zu versuchen, zu verstehen, wie NF-kB-Aktivität zur mitochondrialen Funktion beiträgt. Es wird angenommen, dass der Leser bereits ein Verständnis der grundlegenden mitochondrialen Biologie hat. Im Falle weiterer Studien wird der Leser auf viele hervorragende Studien und Reviews zur mitochondrialen Struktur und Funktion hingewiesen (Hall, 1979; Fox, 1982; Roger und Silberman, 2002; Henze und Martin, 2003; Conradt, 2006; Ettema, 2016; Wang und Youle, 2016; Barshad et al., 2018b).,
NF-kB-Aktivierung
Kernfaktor Kappa B-Untereinheiten, die den NF-kB-Komplex umfassen, werden sowohl in Neuronen als auch in Glia exprimiert. Die NF-kB-Komplex existiert in einem inaktiven Zustand im Zytoplasma (Ghosh et al., 1998; Aggarwal et al., 2004; Hayden und Ghosh, 2004), wo die Aktivierung von NF-kB gut beschrieben wurde (Li und Karin, 2000; Baud und Jacque, 2008; Israel, 2010). Wenn TNFa durch Moleküle wie TNFa oder andere Zellstressoren stimuliert wird, bindet es an TNF-Rezeptoren (Abbildung 1)., Diese Bindung führt über mehrere Zwischenschritte zu einer Wechselwirkung mit dem IkB Kinase (IKK) Komplex, die dann zur Phosphorylierung der IkB führt und anschließend zur Ubiquitination und Degradation der IkB führt. Nach dem Abbau wird das verbleibende NF-kB-Dimer (z. B. p65/p50-Untereinheiten) in den Kern transloziert, wo es an die DNA-Konsenssequenz verschiedener Zielgene bindet. Die Selektivität der NF-kB-Reaktion basiert auf mehreren Faktoren (Sen und Smale, 2010), einschließlich Dimerzusammensetzung, Timing und Zelltyp., Der Einfluss von NF-kB auf das Zellüberleben ist ebenfalls komplex und kann je nach Zelltyp, Entwicklungsstadium und pathologischem Zustand neuroprotektiv oder proinflammatorisch sein (Qin et al., 2007).
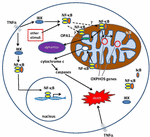
Abbildung 1. Signalwege für den Kernfaktor κ B (NF-kB) im Zytoplasma und im Mitochondrium. Der NF-kB-Tri-Subunt-Komplex (z. B. p65, p50, IkB – eine mögliche Kombination) existiert in einem inaktiven Zustand im Zytoplasma. Die NF-kB-Aktivierung wird ausgelöst, wenn Moleküle wie TNFa an TNF-Rezeptoren binden (verschiedene Typen existieren)., Andere externe oder interne Reize können auch NF-kB aktivieren. Ein komplizierter Signaltransduktionsprozess beginnt dann, sobald TNF-Rezeptoren aktiviert sind; IkB-Kinase (IKK) wird letztlich ausgelöst und führt zur Phosphorylierung der IkB, was zu einer Ubiquitination und Degradation der IkB führt. Sobald IkB abgebaut ist, transloziert das verbleibende NF-kB-Dimer (z. B. p65/p50-oder p50/p50-Untereinheiten-Kombinationen sind möglich) in den Kern, wo es an eine DNA-Konsenssequenz von Zielgenen bindet., Durch Prozesse, die nicht gut verstanden werden, können der NF-kB-Komplex oder NF-kB-Untereinheiten auch in das Mitochondrium wandern, wo Beweise darauf hindeuten, dass es/sie den Intermembranraum einnimmt. Es wird angenommen, dass NF-kB in den Mitochondrien mit OXPHOS-Genen (mitochondrialer mtDNA) interagiert, die zur Expression von Proteinen führen, die an verschiedenen Funktionen beteiligt sind, einschließlich mitochondrialer Dynamik und COX-III-Regulation (Komponente des Komplexes IV)., Es gibt auch Hinweise darauf, dass NF-kB als Schalter in den Mitochondrien fungieren und das Gleichgewicht zwischen der Verwendung von zytoplasmatischer Glykolyse und mitochondrialer Atmung in normalen Zellen und bei Krebs kontrollieren kann. Schließlich deuten die Daten auch auf eine intrinsische apoptotische Signalstimulation hin, bei der die NF-kB-Aktivierung in den Mitochondrien zur Freisetzung von Cytochrom c führt und somit Caspasekaskaden und programmierten Zelltod auslöst.,
Organisatorisch ist NF-kB ein Transkriptionsfaktor der Rel-Familie und ist mit fünf Genen assoziiert, NF-kB1, NF-kB2, RELA, RELB und REL (Chen und Greene, 2004); Diese Gene kodieren für mehrere Proteine, NF-kB1, NF-kB2, RelA, RelB und c-Rel, wobei zwei dieser Proteine große Vorläuferproteine sind, die als p105 und p100 bekannt sind.das wird Proteolyse unterzogen, um p50 und p52 zu werden. Diese Proteine enthalten REL-Homologiedomänen (RHD) in ihrer amino-terminalen Region; Die RHD-Region besteht aus 2 separaten, aber angrenzenden Domänen., Die Sequenz, die am weitesten von der carboxy-terminalen Region entfernt ist, ermöglicht es dem Protein, sich an DNA zu binden. Eine komplexere Sequenz ermöglicht es den Proteinen der Rel-Familie, zur Unterdrückung der Expression über die Bindung ihrer entsprechenden Inhibitorfamilie, der IkB – Proteine, dimerisieren (Homo-oder Heterodimer) (Chen und Greene, 2004). Die letztere Sequenz umfasst die Nuclear-Localization Sequence (NLS), die entlarvt wird, wenn die IkB durch Degradation ungebunden ist. Die NLS hat die Aufgabe, aktive Proteine für den Import in den Zellkern zu führen oder zu markieren (Chen und Greene, 2004; Karin et al., 2004; Barger et al.,, 2005).
Drei dieser Proteine (RelA, RelB und c-REL) kodieren auch eine Transaktivationsdomäne (TADs) in ihrer carboxy-terminalen Region. Die TADs ermöglichen es diesen Proteinen, mit dem Basaltranskriptionsapparat, dem TATA-Bindungsprotein (TBP), dem Transkriptionsfaktor IIB sowie den transkriptionellen Koaktivatoren p300 und cAMP Response Element (CREB) zu interagieren (Chen und Greene, 2004)., Nur diese drei Proteine sind in der Lage, die Transkription ihrer DNA-kodierenden Regionen zu induzieren, während die anderen Proteine, die p50-und p52-Homodimer, die DNA-Bindungsstellen besetzen können, ohne die Transkription zu initiieren. In Anbetracht dessen wirken die späteren 2 Homodimerproteine von p50 und p52 als Transkriptionsrepressoren (Chen und Greene, 2004).
Die Homodimer p105 und p100 besetzen DNA-Bindungsstellen und blockieren so die Transkription über Transkriptionsfaktoren, die TADs besitzen (Barger et al., 2005). Eine dritte Form der transkriptionellen Repression ist die IkB., Diese Proteine haben mehrere Ankyrin-Wiederholungen als Kerndomäne und funktionieren durch Bindung an die RHD, die das NLS maskiert (Karin et al., 2004). Ohne ein aktives NLS sind die NF-kB-Proteine auf das Zytoplasma beschränkt und können nicht in den Kern wandern und so wird die Transkription blockiert.
NF-kB wird 2001 in den Mitochondrien
gefunden, eine Studie von Bottero et al. (2001) fand IkBa und die NF-kB p65-Untereinheit in subzellulären Fraktionen und gereinigten Mitochondrien aus Jurkat-Zellen. Jurkat-Zellen sind eine unsterbliche Zelllinie menschlicher T-Lymphozytenzellen, die zur Untersuchung von Leukämie verwendet werden., In Botteros Studie wurde festgestellt, dass IkBa und NF-kB p65 im mitochondrialen Intermembranraum lokalisiert waren. Der mitochondriale Intermembranraum ist der Raum zwischen der inneren Mitochondrienmembran (IMM) und der äußeren Mitochondrienmembran (OMM).
Anschließend Cogswell et al. (2003) zeigte auch, dass NF-kB-Untereinheiten, p50 und p65 und IkBa, in den Mitochondrien gefunden wurden. Um dies zu bestimmen, wurden verschiedene Methoden verwendet, um Beweise zu liefern, einschließlich Elektronenmikroskopie von Abschnitten von U937-Zellen., U937-Zellen wurden zuerst aus dem Lymphom eines männlichen Patienten mittleren Alters isoliert, um das Verhalten und die Differenzierung von Monozyten zu untersuchen. Hier Cogswell et al. (2003) konnte NF-kB p50-und p65-Untereinheiten und IkBa in der inneren Matrix der Mitochondrien visualisieren. In dieser Studie wurden auch Leberzellen von Ratten untersucht und die Untereinheit p50 und die Untereinheit IkBa identifiziert. Zusätzlich wurden U937-Zellen für 1 h mit TNFa, einem bekannten Auslöser des NF-kB-Signalwegs, stimuliert., In diesem Experiment fanden Western-Blot-Analysen in mitochondrialen und zytoplasmatischen Fraktionen heraus, dass die TNFa-Behandlung einen Verlust von IkBa sowohl im mitochondrialen als auch im zytoplasmatischen Kompartiment um 30 min nach der Behandlung verursachte, was darauf hindeutet, dass IkBa abgebaut wurde. Die EMSA-Analyse, ein In-vitro-Assay, der die NF-kB-Aktivierung und die unspezifische Bindung an DNA-Sequenzen nachweist, wurde auch an Proteinen aus Kernextrakten aus Mitochondrien durchgeführt, die aus mit TNFa stimulierten U937-Zellen isoliert wurden., Hier bestimmten sie TNFa-Signalisierung führte zu einer erhöhten DNA-Bindungsaktivität von NF-kB p50, in Protein aus den Mitochondrien genommen.
Andere Studien haben auch NF-kB in den Mitochondrien nachgewiesen. Dazu gehören Studien (Guseva et al., 2004; Zamora et al., 2004) in humanen Fibroblasten-HT1080-Zelllinien, humanen Prostata-LNCaP-und PC3-Zelllinien und HeLa-Zellen. In LNCaP-Zellen wurden Mitochondrien-NF-kB-p50-und p65-Untereinheiten gefunden, die an mitochondriale DNA (mtDNA) gebunden waren., Zusammengenommen zeigen diese Studien Beweise für die NF-kB-Signalisierung in den Mitochondrien und dass NF-kB die mitochondriale mRNA-Expression reguliert (siehe Abschnitt NF-kB und mitochondriale Genexpression unten).
NF-kB steuert die Mitochondriendynamik
An der Dynamik (Spaltung und Fusion) und Morphologie der Mitochondrien sind mehrere Proteine beteiligt (Karbowski und Youle, 2003; Olichon et al., 2006; Brooks und Dong, 2007; Song et al., 2008; Autret und Martin, 2010; Silva et al., 2013; Sinha und Manoj, 2019). Eine davon ist die Optikusatrophie 1 protein (OPA1) (Olichon et al.,, 2006; Garcia et al., 2018; Lee und Yoon, 2018). Studien haben gezeigt, dass OPA1 ein Regulator der mitochondrialen inneren Membranfusion und auch der mitochondrialen Cristae-Remodellierung ist (Cipolat et al., 2006). Vor kurzem Laforge et al. (2016) zeigte, dass das Fehlen von IKKa einen Einfluss auf die OPA1-Expression in den Mitochondrien und auf die mitochondriale Morphologie hatte.
Überraschenderweise in einer aktuellen Studie von Nan et al. (2017) wurde festgestellt, dass die TNFa-Rezeptor-2 (TNFR2) – Stimulation die mitochondriale Fusion über die NF-kB-abhängige Aktivierung der OPA1-Expression in Herzmyozyten fördert., Wichtig ist, dass die TNFR2-Aktivierung in dieser Studie die Herzmyozyten durch eine Hochregulierung der OPA1-Expression vor Stress schützte. Durch die Verabreichung niedriger Konzentrationen von exogenem TNFa (0,5 ng/ml) vor der Ischämie schien die Reperfusion das Zellüberleben zu verbessern, während höhere Konzentrationen (10-20 ng/ml) zu toxischen Wirkungen in Zellen führten.
NF-kB und Apoptose in den Mitochondrien
Die Rolle der Mitochondrien beim programmierten Zelltod oder Apoptose ist seit geraumer Zeit bekannt (Green und Reed, 1998; Wang und Chen, 2015)., Die wichtigste Rolle für die Mitochondrien ist die Erzeugung von ATP; Die zweitwichtigste Funktion für die Mitochondrien ist jedoch wahrscheinlich die Kontrolle des Zelltods. Wie macht das Mitochondrium das? Wenn das Mitochondrium den Zelltod nicht auslöst, ist Krebs oft die Folge. Um den Zelltod zu regulieren, integrieren Mitochondrien Signale aus einer Vielzahl von Quellen, die als intrinsische Apoptosewege bekannt sind. Komponenten der NF-kB-Aktivität scheinen eines dieser Signale zu sein, obwohl TNFa, ein Aktivator von NF-kB, Teil eines extrinsischen Apoptoseweges ist., Extrinsische Wege (Todesrezeptorvermittelt) werden außerhalb der Zelle eingeleitet, während intrinsische Wege der Apoptose in den Mitochondrien vermittelt und ausgelöst werden.
In einer aktuellen Studie von Pazarentzos et al. (2014) wurde festgestellt, dass IkBa proapoptotische Aktivität ausübt, da es das antiapoptotische NF-kB hemmt. In den meisten Zellen führt die Aktivierung von NF-kB zu einer nachgeschalteten Zielgenexpression, die eine Zelltodresistenz auslöst (Luo et al., 2005). In dieser Studie wurde gezeigt, dass eine neuartige Apoptosefunktion auf IkBa zurückzuführen ist, die Untereinheit, die die Aktivierung von NF-kB hemmt. Pazarentzos et al., (2014) fand heraus, dass IkBa auf das OMM lokalisiert, wo es mit einem spannungsabhängigen Anionenkanal (VDAC) und mitochondrialer Hexokinase II (HKII) interagiert, um diesen Komplex zu stabilisieren und die Bax-vermittelte Cytochrom-c-Freisetzung für Apoptose zu verhindern. Bax ist ein Mitglied der Bcl-2-Familie von Proteinen, von denen gezeigt wurde, dass sie Regulatoren des programmierten Zelltodes sind (Karbowski et al., 2006).
Andere Studien haben auch auf die Rolle von NF-kB bei der direkteren Regulierung der Apoptose im Mitochondrium hingewiesen. In einer Studie von Liu et al., (2004) führte die Hemmung von NF-kB allein in Makrophagen zur Freisetzung von Cytochrom c. Denken Sie daran, dass Cytochrom c für das Pendeln von Elektronen von Komplex III nach Komplex IV verantwortlich ist und dass die Freisetzung von Cytochrom c in das Zytoplasma, einen Aktivator von Caspasen, ein Schlüsselschritt bei der Auslösung von Apoptose ist.
NF-kB und mitochondriale Atmung
Nuklearer Faktor Kappa B wurde in vielen Studien zur Förderung der Tumorigenese gezeigt. Wie dies geschieht, war nicht genau klar. In einer bahnbrechenden Studie von Mauro et al., (2011) wurde festgestellt, dass NF-kB die mitochondriale Atmung in Kolonkarzinomzellen hochreguliert. Hier stellten sie fest, dass diese Funktion von NF-kB den Warburg-Effekt unterdrückt. Daran erinnern, dass der Warburg-Effekt (Vander Heiden et al., 2009) beschreibt die Beobachtung, dass Krebszellen den Stoffwechsel eher durch Glykolyse als durch den effizienteren oxidativen Phosphorylierungsweg begünstigen. In dieser Studie stellten die Autoren fest, dass NF-kB Netzwerke des Energiestoffwechsels organisiert, indem das Gleichgewicht zwischen Glykolysenutzung und mitochondrialer Atmung kontrolliert wird., Interessanterweise fanden sie eine Rolle für NF-kB bei der Stoffwechselanpassung in normalen Zellen und bei Krebs. Ihre Ergebnisse deuteten ferner darauf hin, dass die Unterdrückung des mitochondrialen Stoffwechsels in etablierten Krebszellen durch Hemmung von NF-kB und Metformin die Tumorigenese verringert.
NF-kB und mitochondriale Genexpression
Kernfaktor Kappa B ist ein bekannter Regulator der Genexpression-sowohl negativ als auch positiv (Mattson et al., 2000). Wie NF-kB die nuklear kodierte mitochondriale Genexpression reguliert oder beeinflusst, ist jedoch weniger bekannt., Menschliche mtDNA besitzen 37 Gene, die für 13 Polypeptide kodieren. Es wurde gezeigt, dass mtDNA-Gene für viele der Untereinheiten aller 5 Komplexe der Elektronentransportkette (ETC), 2 rRNAs und 22 tRNAs codieren. Die meisten ETC-Untereinheiten sind jedoch durch Kern-DNA codiert, die durch NF-kB-Aktivität beeinflusst werden könnte (Calvo et al., 2016).
Zum Beispiel wurde behauptet (Cogswell et al., 2003), dass der NF-kB-Weg die mitochondriale Genexpression, die mit der COX III-Untereinheit assoziiert ist, negativ regulieren kann., Die COX III-Untereinheit wird durch mtDNA codiert und ist Bestandteil des Komplexes IV im mitochondrialen SYSTEM. Es fungiert als katalytische Untereinheit im Komplex IV, dem Komplex, der mit dem mitochondrialen Sauerstoffverbrauch verbunden ist. In einer Studie von Cogswell et al. (2003) führte die Modulation der NF-kB-Aktivierung zum Verlust der Expression von COX III-und Cytochrom-b-mRNA. Andere Studien unterstützen eine Rolle für NF-kB, die zusätzliche mitochondriale Gene wie COX I und Cytb regulieren (Psarra und Sekeris, 2008, 2009; Barshad et al., 2018b)., Zusätzlich reduzierte die NF-kB-p65-Untereinheit die Spiegel von mtDNA-kodierter CytB-mRNA, möglicherweise durch Bindung an die D-Schleife in menschlichen Zellen in Abwesenheit von p53 (Johnson et al., 2011). Insgesamt deuten diese Ergebnisse darauf hin, dass die NF-kB-Signalisierung die enzymatische Aktivität von respiratorischen ETC-Komplexen beeinflussen kann.
NF-kB vermittelt eine Aß-induzierte Dysfunktion in den Mitochondrien
Die Alzheimer-Krankheit (AD) ist mit dem Aufbau von Aß-Plaques und/oder dem Auftreten von neurofibrillären Verwicklungen (NFTs) in bestimmten Hirnregionen verbunden (Duyckaerts et al., 2009)., Es gibt jedoch Kontroversen darüber, ob Aß ein Erreger von AD ist oder ob Aß einfach mit dem Altern korreliert. Akkumulieren von Beweisen (Aliev et al., 2009; Correia et al., 2012; Cadonic et al., 2016; Cardoso et al., 2017; Djordjevic et al., 2017) weist nun auch auf Veränderungen des Hirnstoffwechsels hin, die durch mitochondriale Dysfunktion als einen Prozess verursacht werden, der für viele altersbedingte neurodegenerative Störungen, einschließlich AD, von zentraler Bedeutung ist. Hinzu kommen Beeinträchtigungen der enzymatischen Aktivität der Proteinkomplexe des ETC und Veränderungen der antioxidativen enzymatischen Aktivität (Kolosova et al.,, 2017) in AD. Insbesondere hat sich gezeigt, dass die komplexe IV-Aktivität bei AD negativ beeinflusst wird (Mutisya et al., 1994).
In einer aktuellen Studie von Shi et al. (2014) wurde festgestellt, dass Aß die Mitochondrienfunktion über die NF-kB-Signalisierung beeinträchtigte. Darüber hinaus Shi et al. (2014) zeigte hier, dass Aß die Expression der COX III-Untereinheit über einen NF-kB-Weg verringerte., Um die Möglichkeit zu eliminieren, dass IkBa durch Aß im Zytoplasma phosphoryliert (und dann in die Mitochondrien transportiert) wurde, wurden isolierte Mitochondrien in Gegenwart (oder Abwesenheit) eines NF-kB-Blockers, nämlich BAY11-7082, mit Aß inkubiert. Hier fanden Sie Aß-induzierte Phosphorylierung und degradation von IkBa in isolierten Mitochondrien.
Diese Ergebnisse haben auch wichtige Implikationen für die AD-Behandlung, wie jüngste Studien von Snow et al. (2018) und wie in anderen verwandten Studien gezeigt (Djordjevic et al., 2017; Adlimoghaddam et al.,, 2019), die darauf hindeuten, dass die NF-kB-Signalisierung in den Mitochondrien einen therapeutischen Wert haben kann. Zum Beispiel, im Schnee et al.es wurde gezeigt, dass Kreatin – ein bekannter Modulator der Mitochondrienfunktion (Tarnopolsky und Beal, 2001)-die Proteinspiegel von CaMKII, PSD-95 und Complex 1-Untereinheiten in kreatingefütterten Mäusen erhöht und positiv verändert, während die NF-kB-inhibitorische IkB-Untereinheit verringert wurde. Weitere Informationen zur potenziellen therapeutischen Wirkung von Kreatin auf die Mitochondrienfunktion und bei mitochondrialen Störungen oder anderen neurologischen Störungen finden Sie in Studien und Reviews von Matthews et al., (1998), Klivenyi et al. (1999), Tarnopolsky und Beal (2001), Hersch et al. (2006), Rodriguez et al. (2007) und Beal (2011).
NF-kB Rolle bei Entzündungen und mitochondrialen Stoffwechsel
Zunehmende Daten (Lamas et al., 2003; Mauro et al., 2011; Moretti et al., 2012) deuten darauf hin, dass die NF-kB-Signalisierung, die ein Mediator entzündlicher Prozesse ist, auch als Regulator und Integrator des Energiestoffwechsels fungiert. In einer aktuellen Studie von Zhong et al. (2016) wurde gezeigt, dass NF-kB die Aktivierung des Inflammasoms durch Eliminierung geschädigter Mitochondrien einschränkt., Überraschenderweise schien NF-kB sowohl das NLRP-3-Inflammasom für die Aktivierung zu primieren als auch eine übermäßige Entzündung zu verhindern und die Aktivierung des NLRP-3-Inflammasoms zu hemmen; obwohl der Mechanismus für die Zurückhaltung schlecht definiert war. Hier wurde spekuliert, dass NF-kB nicht nur ein Aktivator entzündlicher Gene ist, sondern auch in dieser Studie durch Begrenzung der NLRP3-Inflammasomaktivierung und IL-1β-Produktion funktioniert. Darüber hinaus wurde festgestellt, dass die p62-Induktion für die Inflammasom-inhibitorische Aktivität durch NF-kB verantwortlich war., Es scheint, dass NF-kB seine eigene Entzündung in Makrophagen zurückhalten kann, indem es die p62-vermittelte Entfernung geschädigter Mitochondrien (Mitophagie) fördert, nachdem Makrophagen mit verschiedenen NLRP3-Inflammasomaktivatoren interagieren.
Schlussfolgerung
Vor über 10 Jahren wurde NF-kB in den Mitochondrien nachgewiesen. Überraschenderweise wurden für einen so wichtigen Transkriptionsfaktor wenig Fortschritte bei der Aufdeckung spezifischer Rollen für NF-kB erzielt, die das Mitochondrium beeinflussen., Einige Studien, wie oben beschrieben, liefern Hinweise auf NF-kB in der Mitochondriendynamik, Apoptose, Atemkontrolle, Genexpression und Krankheitsmechanismen (Abbildung 1). Eine Vervielfältigung dieser Ergebnisse und eine allgemeine Validierung durch andere Laboratorien sind jedoch weiterhin erforderlich. Einige zusätzliche Erkenntnisse können aus der Tatsache gewonnen werden, dass andere Transkriptionsfaktoren, die Auswirkungen auf Kerngene haben, wie AP-1, p53, CREB, c-Myc, Wnt13, Dok-4, HMGA1 und c-Src, auch in den Mitochondrien nachgewiesen wurden (Psarra und Sekeris, 2008)., Interessanterweise wurden Bindungsstellen im mitochondrialen Genom (homolog zu ihren Bindungsstellen in der Kern-DNA) für einige dieser Faktoren bestimmt (Psarra und Sekeris, 2008), bei denen Rollen für mitochondriale Transkription und Apoptose vermutet werden und einige allgemeine Aktivitätsmuster zeigen. Beispielsweise kann argumentiert werden, dass einige dieser Faktoren (NF-kB, CREB und AP-1) an mitochondriale Genome binden und meist die mitochondriale Genexpression abschwächen (Blumberg et al., 2014), während stimulierende Wirkungen auf die nukleare Gentranskription haben., Es ist jedoch deutlich mehr Arbeit erforderlich, um nicht nur genaue Rollen der Aktivität zu finden, sondern auch festzustellen, ob die gesamten Aktivitätsmuster wirklich existieren.
Nach der Untersuchung dieser Literatur wird auch deutlich, dass die Rolle von NF-kB bei der Regulation der mitochondrialen Atmung tiefgreifende Auswirkungen hat und ein Ausmaß an Komplexität aufweist, das bisher nicht geschätzt wurde. Zum Beispiel, Mauro et al. (2011) Daten legen eine Rolle für NF-kB bei der Stoffwechselanpassung in normalen Zellen und bei Krebs fest und deuten auch auf Konsequenzen für andere Krankheitszustände wie AD hin., Darüber hinaus kann NF-kB seine eigene Entzündung zurückhalten, wie von Zhong et al. (2016) ist nicht nur überraschend, sondern veranschaulicht auch die Komplexität der NF-kB-Signalisierung in der mitochondrialen Funktion.
In diesem review wurden Studien Befragten auf NF-kB Rolle in der mitochondrialen Funktion, und es scheint, dass die Forschung in diesem Bereich ist Steigend. Erschwerend für die Ergebnisse ist jedoch die Beobachtung, dass mehrere Faktoren eine ähnliche Rolle in der Mitochondrienfunktion spielen und daher detaillierte Studien erforderlich sind, die für jeden Faktor spezifisch sind., Abschließend können wir die Frage erneut stellen-Was macht NF-kB im und mit dem Mitochondrium? Die sofortige und abgekürzte Antwort wäre-viel!
Autorenbeiträge
Der Autor hat das Thema erstellt und das Manuskript geschrieben.
Finanzierung
Diese Arbeit wurde durch Fördermittel der Canadian Institutes of Health Research (CIHR), der Canadian Agricultural Partnership (CAP), der St. Boniface Hospital Research Foundation, der Alzheimer Society of Manitoba, der Research Manitoba, der Honourable Douglas und Patricia Everett sowie des Royal Canadian Properties Limited Endowment Fund unterstützt.,
Erklärung zum Interessenkonflikt
Der Autor erklärt, dass die Untersuchung ohne kommerzielle oder finanzielle Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagungen
Der Autor möchte Dr. Grant Hatch für eine Überprüfung des Manuskripts danken.
Hayden, MS und Ghosh, S. (2004). Signalisierung an NF-kappaB. Gene Dev. 18, 2195–2224.
PubMed Abstract | Google Scholar
Wang, X. Chen, X. J. (2015)., Ein zytosolisches Netzwerk, das Mitochondrien-vermittelten proteostatischen Stress und Zelltod unterdrückt. Nature 524, 481-484. doi: 10.1038 / nature14859
PubMed Abstract / CrossRef Volltext / Google Scholar