Introduction
L’achèvement du projet sur le génome humain a été une étape importante pour les connaissances médicales, fournissant les informations nécessaires à la compréhension des caractéristiques uniques de chaque individu.1 la conséquence logique de ces connaissances serait de pouvoir appliquer des tests diagnostiques et des traitements spécifiques à chaque patient sur la base de leurs informations génétiques individuelles. Cette nouvelle forme de soins médicaux est appelée médecine personnalisée.,2 Cependant, malgré les grands progrès qui ont conduit à la connaissance du génome humain, sa traduction en traitements diagnostiques et personnalisés a été moins que prévu. À l’heure actuelle, deux initiatives majeures vont dans ce sens: la biologie des systèmes3 et la pharmacogénétique.,4 le but ultime de ces initiatives est de développer une pratique médicale adaptée aux caractéristiques de chaque individu qui peut prédire l’apparition ou l’évolution d’une maladie particulière, permettre l’établissement de stratégies de prévention appropriées et, enfin, permettre au patient de prendre part à la prise de décision. Cela a été appelé médecine P4.5
la fibrose kystique (FK) reste la maladie génétique la plus fréquente et la plus mortelle chez les Caucasiens., Il se produit à un taux de 1 nouveau-né sur 2500-6000, selon la région et l’origine ethnique, et dans une proportion de porteurs sains, variant entre 1:20 et 37.6 en Espagne, grâce à l’introduction progressive de programmes de dépistage néonatal dans les différentes communautés, une incidence de mucoviscidose est maintenant plus faible que ce qui était estimé précédemment en 2009, soit 1/4430 naissances vivantes en Galice, 1/4339 en Castille-Leon, 1/5376 à Murcie, 1/5840 en Catalogne et 1/6602 aux Baléares.7 on estime qu’il y a 70 000 patients atteints de mucoviscidose dans le monde.,8 la maladie est causée par des mutations dans le gène codant pour la protéine régulatrice du régulateur de conductance transmembranaire de la fibrose kystique (CFTR), un canal chlorure impliqué dans la libération de l’adénosine triphosphate et la régulation d’autres canaux de transport d’ions. Cette protéine est exprimée dans les cellules épithéliales respiratoires, le pancréas, les voies biliaires, les glandes sudoripares et le système génito-urinaire., Son altération entraîne une anomalie dans le transport des ions, de sorte que les patients produisent un mucus épais et collant qui obstrue les canaux de l’organe où il se trouve et que l’altération présente des effets multisystémiques qui déterminent le large éventail de manifestations cliniques de la mucoviscidose. Malgré des avancées majeures dans le traitement de la mucoviscidose qui ont entraîné une survie plus longue (la médiane actuelle est estimée à 37,5 ans) 9,Il reste encore beaucoup de chemin à PARCOURIR pour que les patients atteints de mucoviscidose aient une quantité et une qualité de vie similaires à celles des sujets non atteints de la maladie., Dans ce contexte, de nouveaux traitements pour diminuer la morbidité et augmenter la survie sont nécessaires.
la FC est un exemple de maladie bien placée pour tirer parti de la médecine personnalisée. D’une part, c’est une maladie monogénique, causée par des mutations dans un gène spécifique. La physiopathologie de l’entité est bien caractérisée et les cibles thérapeutiques sont claires. En outre, le diagnostic de la maladie nécessite des tests génétiques pour l’identification du type de maladie, de sorte que le défaut génétique exact est déterminé dans chaque cas.,10
à l’heure actuelle, deux approches très différentes visent à corriger le défaut fondamental: la thérapie génique, visant à corriger l’altération génétique, et la thérapie moléculaire, visant à corriger le défaut fonctionnel au niveau des protéines. L’objectif de la thérapie génique est l’introduction de copies génétiques normales dans les voies respiratoires des patients atteints de mucoviscidose. Elle implique l’insertion d’un vecteur viral recombinant, dont l’ADN a été extrait et remplacé par le nouvel ADN thérapeutique. Le vecteur viral Sert de véhicule pour insérer le nouvel ADN dans la cellule cible., Divers types de virus, tels que l’adénovirus ou le lentivirus, ont été utilisés à ce jour. En outre, des particules non virales, telles que des nanoparticules capables d’insérer de l’ADN, ont également été développées.11 cependant, les résultats jusqu’à présent ont été médiocres, car la durée d’expression du gène introduit était courte avec les deux types de vecteurs.12 Le Consortium britannique de thérapie génique développe un essai clinique de phase II pour évaluer l’efficacité clinique d’un vecteur d’ADN plasmidique/liposomique optimisé.13 l’objectif de recrutement est de 130 patients et les résultats sont attendus en 2014 (NCT01621867).,14
d’autre part, la thérapie visant à restaurer la fonction de la protéine CFTR a été plus réussie. Ces dernières années, les résultats commencent à venir à travers les médicaments qui agissent directement sur la protéine CFTR. En fait, en janvier 2012, le premier médicament pour corriger les défauts de mutation Gly551Asp a été commercialisé aux États-Unis. Dans les sections suivantes, nous passerons en revue les informations disponibles sur les progrès de la médecine personnalisée pour la mucoviscidose et les traitements disponibles visant à corriger le défaut causant la maladie au niveau des protéines., Dans cette revue, La nomenclature utilisée pour la description des mutations du gène CFTR développée par la Human Genome Variation Society15 sera utilisée.
Mutations et défaut protéique
la FC est une maladie héréditaire autosomique récessive, de sorte que la mutation doit être présente dans les deux copies du gène CFTR pour être affectée. À ce jour, plus de 1900 mutations du gène CFTR associées à la maladie ont été identifiées dans la séquence codante, L’ARN messager ou d’autres éléments. Les Mutations du gène CFTR sont disponibles pour consultation dans la base de données sur les mutations de la fibrose kystique.,16 la première mutation décrite, et la plus courante dans le monde, est Phe508del, mais il existe d’autres mutations spécifiques dont la fréquence varie selon les groupes ethniques. En Espagne, la fréquence moyenne de la mutation Phe508del est comprise entre 50% et 60% de tous les chromosomes étudiés, le deuxième plus fréquent est Gly542X avec 4% -8%, suivi de Asn1303Lys dans 2% -4% des cas. Les mutations décrites à ce jour sont classées en six types ou classes selon le mécanisme à l’origine de la maladie.17 Ces types de mutations sont résumés à la Fig. 1., Les mutations de classe I conduisent à un codon d’arrêt prématuré dans l’ARN messager qui empêche la traduction de la protéine complète. Ainsi, la protéine produite est courte et non fonctionnelle. Les mutations de classe II codent une protéine structurellement anormale et mal repliée qui est éliminée par le réticulum endoplasmique avant d’atteindre la surface cellulaire. La mutation la plus fréquente dans la FC, Phe508del, appartient à ce groupe. Dans le cas de mutations dans les classes III à VI, les protéines atteignent la surface cellulaire, mais ne fonctionnent pas correctement. Les mutations de classe III entraînent une diminution de l’activation des canaux, de sorte que les canaux restent fermés., Les mutations de classe IV provoquent une diminution de la conductance ionique à travers le canal. Les mutations de classe V codent des protéines mineures entraînant une quantité réduite de CFTR dans la surface cellulaire, de sorte qu’une certaine fonction se produit, mais à un niveau réduit. Enfin, les mutations de classe VI entraînent une demi-vie raccourcie en raison de l’instabilité des protéines et peuvent également endommager la régulation des canaux CFTR voisins à la surface cellulaire.
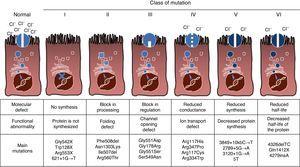
types de mutations dans la fibrose kystique.
CFTR Modulateurs
Trois classes principales ont été identifiées dans le développement de médicaments pour la réparation de la protéine CFTR.11 le premier groupe sont les suppresseurs de codon d’arrêt prématuré (mutations de classe I). Ces médicaments empêchent l’identification de ce codon d’arrêt prématuré, de sorte que la synthèse des protéines peut se poursuivre jusqu’à la fin. Le deuxième groupe sont les correcteurs CFTR. Ces composés sont conçus pour corriger les défauts dans le transport de la protéine pliée (mutations de classe II) vers la membrane cellulaire, où elle peut fonctionner presque normalement., Le troisième groupe est constitué des potentialisateurs dits CFTR. Ce sont des médicaments conçus pour cibler la protéine CFTR à la surface de la cellule afin d’améliorer sa fonction. Ainsi, ces potentiateurs peuvent agir sur les mutations de classe III, IV, V et VI. Actuellement, de nombreuses molécules utilisant ces différents mécanismes sont à l’étude, dont L’une a déjà atteint le marché: Ivacaftor (VX-770) est un potentiateur CFTR approuvé aux États-Unis en janvier 2012 pour le traitement des patients atteints de mucoviscidose âgés de plus de 6 ans et porteurs de la mutation Gly551Asp.,
traitements par Mutation de classe I
Environ 10% des cas de mucoviscidose sont causés par des mutations de classe I. Les premiers médicaments utilisés pour cette classe étaient les aminoglycosides. Il y a plusieurs années, on a rapporté que la gentamicine avait la capacité de masquer le codon d’arrêt prématuré empêchant la synthèse de la protéine CFTR. Ceci est réalisé par l’insertion d’un acide aminé qui permet au ribosome de continuer à lire le gène, produisant une protéine pleine longueur. Des études précliniques ont démontré que la protéine pouvait être synthétisée dans un modèle murin et que 35% de la fonction protéique était récupérée in vitro.,18,19 L’effet de l’administration intraveineuse de gentamicine a été évalué dans deux études de patients atteints de mucoviscidose présentant différents types de mutations de classe I. L’un a été mené aux États-Unis chez cinq patients,20 et un autre comprenait 18 patients en France.21 cependant, bien que la réponse ait été positive, les résultats variaient considérablement, de sorte que l’avantage n’était pas universel. De plus, les problèmes de toxicité des aminoglycosides ont contribué à un profil défavorable.
Une alternative synthétique est l’ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., Il s’agit d’une molécule conçue pour permettre aux ribosomes de lire l’information génétique tout en « sautant” le codon d’arrêt prématuré, produisant une protéine CFTR fonctionnelle.22 La pharmacocinétique de l’ataluren a été démontrée dans des modèles animaux et dans des essais de phase II. Dans deux petites études préliminaires, 23, 24 un groupe de patients atteints de mucoviscidose traités par ataluren oral a montré une amélioration des anomalies électrophysiologiques de la maladie, avec une augmentation du nombre de cellules du nez exprimant la protéine à leur surface., Par la suite, une autre petite étude de 19 patients a évalué différentes doses d’ataluren administrées par voie orale toutes les 8h, avec une amélioration de l’activité du CFTR et des paramètres cliniques et un bon profil de sécurité.25 les résultats d’un essai clinique de phase III de l’ataluren qui n’avaient pas encore été officiellement publiés ont été rapportés lors de la Conférence nord-américaine sur la fibrose kystique de 2012. Au total, 238 patients âgés de plus de 6 ans ont été randomisés pour recevoir 10, 10, 20 mg/kg d’ataluren ou un placebo toutes les 8h pendant 48 semaines., Après 48 semaines de traitement, il n’y a pas eu de différence significative entre le VEF1 de l’ataluren et le placebo (-2,5% d’ataluren vs -5,5% de placebo, P=ns). Une fois stratifiée par l’utilisation chronique d’antibiotiques nébulisés, il y avait une différence de 6,7% dans le changement moyen après 48 semaines en faveur de l’ataluren pour les patients ne prenant pas de tobramycine, alors qu’il n’y avait aucune différence dans le changement de FEV1 chez ceux recevant de la tobramycine nébulisée. Quelque chose de similaire s’est produit avec le pourcentage d’exacerbations, où la différence entre les deux groupes était significative., Une fois stratifiés, les patients du groupe ataluren non sous tobramycine nébulisée ont montré une diminution en pourcentage des exacerbations de 43% par rapport au groupe placebo. Les tests de différence de potentiel Nasal et de chlorure de sueur n’ont montré aucune différence entre les groupes, indépendamment de l’utilisation d’antibiotiques nébulisés. Les auteurs ont conclu que les avantages étaient plus importants chez les patients ne recevant pas d’antibiothérapie chronique avec un aminoglycoside nébulisé, spéculant que la tobramycine et l’ataluren interagissaient à un niveau ribosomique, produisant un antagonisme lorsqu’ils étaient utilisés simultanément.,26
traitements par Mutation de classe II
Les mutations de classe II, étant la mutation la plus fréquente de la maladie (Phe508del), sont présentes chez un grand nombre de patients atteints de mucoviscidose, ce qui en fait une cible principale dans la recherche sur la mucoviscidose. Diverses molécules ont été étudiées, dont la plupart ont été développées par Vertex Pharmaceuticals Inc. Le premier composé correcteur, lumacaftor (VX-809), a montré une bonne efficacité in vitro, améliorant le transport du chlorure dans 14% des cas.,27 cependant, les résultats ont été quelque peu décevants chez les patients, car les améliorations de la concentration de chlorure dans la sueur étaient très faibles (7mmol/l) et il n’y avait aucun changement dans le potentiel nasal.28
Les effets du potentiateur CFTR ivacaftor (VX-770), un médicament pour les mutations de classe III (voir description ci-dessous), ont également été étudiés chez des patients homozygotes pour Phe508del. Les résultats de L’étude DISCOVER montrent que l’ivacaftor n’est pas associé à une amélioration significative du VEF1, de la qualité de vie ou du nombre d’exacerbations par rapport au placebo.,29
étant donné que le lumacaftor peut aider à l’administration de Phe508del-CFTR à la surface cellulaire et que l’ivacaftor augmente le temps d’ouverture et la conduction du chlorure à travers la cellule épithéliale, l’amélioration du défaut sous-jacent de Phe508del peut être possible avec la combinaison des deux molécules. Des études in vitro d’ivacaftor et de lumacaftor combinés dans des épithéliums respiratoires avec mutation Phe508del ont montré que le lumacaftor augmente seul le transport du chlorure CFTR de 15%, et lorsque l’ivacaftor est ajouté, le transport augmente à près de 30%., Cette combinaison de médicaments a été étudiée dans une étude de phase II chez des patients présentant une mutation Phe508del. Les résultats complets n’ont pas encore été publiés, mais les données initiales suggèrent un effet bénéfique sur la fonction pulmonaire dans le phe508del homozygote, mais pas dans l’hétérozygotie.30 deux essais cliniques chez des patients de 12 ans ou plus homozygotes pour la mutation Phe508del sont en cours de développement pour évaluer l’ivacaftor et le lumacaftor combinés; il s’agit des études TRAFFIC (NCT01807923)31 et TRANSPORT (NCT01807949)32. Les résultats pourraient permettre à environ la moitié de la population de FC de recevoir un traitement modulant CFTR., Cependant, des études sont nécessaires pour évaluer l’effet de la thérapie combinée chez les individus hétérozygotes Phe508del.
Une autre alternative est le composé correcteur VX-661, et des études sont actuellement en cours. Son efficacité est testée seule et en association avec l’ivacaftor (VX-770) et les résultats seront bientôt disponibles (NCT01531673).33
traitements de Mutation de classe III
L’Ivacaftor (VX-770) est un potentialisateur CFTR qui module la fonction de la protéine anormale.,34 cette molécule a été initialement conçue pour améliorer la fonction CFTR dans des cultures de cellules épithéliales respiratoires porteuses d’une seule mutation Gly551Asp.34 c’est le premier médicament approuvé aux États-Unis et en Europe pour le traitement de la mucoviscidose chez les patients porteurs de la mutation Gly551Asp. Après avoir démontré in vitro la capacité de l’ivacaftor à améliorer le transport du chlorure à travers la membrane cellulaire, les premiers essais de phase II ont été menés auprès de 39 patients.35 dans cette étude, la fonction de la protéine CFTR s’est améliorée trois jours après le début du traitement, atteignant des concentrations de chlorure dans la sueur allant jusqu’à des niveaux normaux., Avec ces résultats, deux autres études cliniques ont suivi: L’étude STRIVE, chez 144 patients âgés de 12 ans OU plus36 et L’étude ENVISION incluant 52 enfants âgés de 6 à 11 ans.37 les deux études comprenaient des patients présentant au moins une mutation Gly551Asp et un VEF1 compris entre 40% et 105% qui ont été initialement suivis pendant une période de 14 jours, puis randomisés pour recevoir 150 mg d’ivacaftor oral ou un placebo deux fois par jour pendant une période de 48 semaines., Après avoir terminé 48 semaines de traitement, les patients ont eu la possibilité de poursuivre une étude longitudinale ouverte, L’étude PERSIST (nct01117012),38 pendant 96 semaines.
dans STRIVE, les patients du groupe ivacaftor ont présenté une amélioration de 10,6% du VEF1 (critère d’évaluation primaire) à partir du 15e jour de traitement, qui a été maintenue au cours de l’étude de 48 semaines. De plus, une diminution de la concentration de chlorure dans la sueur a été observée (moyenne: -48,7 mmol/L), avec une amélioration de la qualité de vie, une réduction de 55% des exacerbations et une prise de poids de 2,7 kg.,
les résultats de L’étude ENVISION coïncident en grande partie avec ceux de l’étude chez les adolescents et les adultes, avec la différence que la qualité de vie n’a pas atteint la différence statistique. Les effets indésirables les plus fréquemment observés dans le groupe de traitement dans les études STRIVE et ENVISION ont été des infections des voies respiratoires supérieures, une congestion nasale, des maux de gorge, des étourdissements et des éruptions cutanées., Les résultats préliminaires après les 12 premières semaines de L’étude persistent révèlent que l’amélioration de la fonction pulmonaire (VEV1), des symptômes respiratoires et de la prise de poids chez les patients traités par ivacaftor est maintenue pendant cette période. De plus, le sous-groupe de patients qui sont passés du placebo à l’ivacaftor à L’inclusion dans PERSIST a connu une amélioration de 10,8% du VEF1 à 15 jours et de 13% à 12 semaines, ainsi qu’une réduction des exacerbations.,39
malgré les bons résultats obtenus avec l’ivacaftor dans le traitement de la mutation Gly551Asp chez les enfants de plus de 6 ans et les adultes à 48 semaines, certains problèmes restent à résoudre. Premièrement, le médicament n’a pas été testé chez les enfants de moins de 6 ans. Cependant, il semble judicieux de corriger le défaut avant que des dommages irréversibles ne se produisent, étant donné que l’atteinte pulmonaire commence avant l’âge de six ans. Un essai dans cette tranche d’âge est actuellement en cours (NCT01705145).40 deuxièmement, une autre alternative serait d’essayer également d’autres mutations de classe III., À cet égard, des études in vitro sur neuf autres mutations ont montré des résultats très similaires41, il est donc raisonnable de s’attendre à des résultats similaires chez les patients. Un essai clinique de phase III est en cours chez des patients âgés de plus de 6 ans présentant d’autres mutations de classe III (études KONTINUE et KONNECTION; nct01614470).42,43 Troisièmement, bien qu’aucun traitement ne soit disponible pour le reste des classes de mutation (IV–VI), les potentialisateurs CFTR peuvent être également bénéfiques dans ces cas., Un essai clinique est en cours à cet égard, évaluant l’efficacité de l’ivacaftor dans la mutation Arg117His de classe IV (étude KONDUCT; NCT01614457).44 Enfin, l’efficacité et l’innocuité à long terme de l’ivacaftor au-delà de 48 semaines n’ont pas encore été établies. L’étude observationnelle G551D (GOAL; NCT01521338) 45 est une étude observationnelle suivant des patients âgés de plus de 6 ans recevant de l’ivacaftor., Il vise à rendre compte de l’efficacité et de l’innocuité de l’ivacaftor à long terme, ainsi que d’autres résultats intéressants, notamment les médiateurs inflammatoires dans les expectorations, la clairance mucociliaire et le pH gastro-intestinal. les résultats sont attendus à la fin de 2013.
Conclusions
la FC est un exemple de maladie bien placée pour tirer parti de la médecine personnalisée. À l’heure actuelle, deux approches très différentes visent à corriger le défaut de base: la thérapie génique visant à corriger l’altération génétique, et la thérapie avec des molécules visant à corriger le défaut fonctionnel au niveau des protéines., Ce dernier commence à montrer des résultats prometteurs pour diverses molécules en développement, et l’une d’entre elles (ivacaftor) est déjà commercialisée pour la mutation Gly551Asp classe III, avec d’excellents résultats chez les enfants de plus de 6 ans, les adolescents et les adultes. L’objectif final est de fournir des traitements Correcteurs et potentialisateurs pour tous les patients atteints de mucoviscidose quelle que soit leur mutation. Comme les résultats de ces et d’autres nouvelles molécules apparaissent, il est probable qu’une molécule ou combinaison sera nécessaire pour chaque patient, en fonction de leurs mutations., Dans tous les cas, l’avenir est prometteur et des mesures importantes ont été prises pour obtenir un traitement qui agira efficacement sur la cause de cette maladie.
conflits d’intérêts
Les auteurs ne déclarent aucun conflit d’intérêts.