Johdanto
päätökseen human genome project oli merkityksellinen virstanpylväs lääketieteen tietämystä, joka tarjoaa tiedot, jotka ovat tarpeen ymmärrystä ainutlaatuisia ominaisuuksia kunkin.1 looginen seuraus tästä tietoa, olisi voitava soveltaa erityisiä diagnostisia testejä ja hoitoja kullekin potilaalle, joka perustuu niiden yksilölliset geneettiset tiedot. Tätä uutta sairaanhoidon muotoa kutsutaan personoiduksi lääketieteeksi.,2 Kuitenkin, vaikka suurta edistystä, joka on johtanut tietoa ihmisen perimästä, sen käännös diagnostisia ja yksilöllisiä hoitoja on ollut odotettua vähemmän. Tällä hetkellä tähän suuntaan on menossa kaksi merkittävää aloitetta: systems biology3 ja pharmacogenetics.,4 perimmäinen tavoite, että näitä aloitteita on kehittää lääketieteen käytännössä räätälöity ominaisuudet kunkin yksittäisen, joka voi ennustaa puhkeamista tai tietyn sairauden, antaa asianmukaisia ennalta ehkäiseviä strategioita laaditaan ja lopuksi, jotta potilas voi osallistua päätöksentekoon. Tätä on kutsuttu P4-lääkkeeksi.5
Kystinen fibroosi (CF) on yleisin ja tappava geneettinen sairaus Valkoihoisilla., Se tapahtuu nopeudella 1 2500-6000 vastasyntyneet, riippuen alueesta ja etnisestä alkuperästä, ja osuus on terveitä kantajia, vaihdellen välillä 1:20 ja 37,6 Espanjassa, kiitos progressiivinen käyttöönotto vastasyntyneiden seulonta ohjelmia eri yhteisöjen, harvemmin CF on nyt nähnyt kuin aiemmin oli arvioitu vuonna 2009, eli 1/4430 elävänä syntyneiden Galiciassa, 1/4339 Castilla-Leon, 1/5376 Murcia, 1/5840 Kataloniassa, ja 1/6602 baleaareilla.7 CF: stä kärsiviä arvioidaan olevan maailmanlaajuisesti 70000.,8 sairaus johtuu mutaatioista geeni, joka koodaa sääntelyn proteiinia kystinen fibroosi läpäisevä johtokyky säädin (CFTR), a chloride channel mukana-julkaisu adenosiinitrifosfaatin ja asetuksen muita ion liikenteen kanavat. Tämä proteiini ilmaistaan hengitysteiden epiteelisoluissa, haimassa, sappiteissä, hikirauhasissa ja urogenitaalijärjestelmässä., Sen muuttaminen johtaa vamman ion transport, niin että potilaat tuottavat paksu, tahmea lima, joka tukkii kanavat elin, jossa se sijaitsee ja niin muutos esittelee vaikutuksia useisiin elinjärjestelmiin, jotka määrittävät erilaisia kliinisiä ilmenemismuotoja VRT. Vaikka merkittäviä edistysaskeleita hoitoon CF, joka on johtanut pidempään selviytyminen (nykyinen mediaani on arvioitu 37,5 vuotta),9 on vielä pitkä matka varmistaa, että potilailla, joilla CF on määrä ja laatu elämän muistuttavat, että tutkittavien ilman tauti., Tässä yhteydessä tarvitaan uusia hoitoja sairastavuuden vähentämiseksi ja eloonjäämisen lisäämiseksi.
CF on esimerkki sairaudesta, jolla on hyvät mahdollisuudet hyödyntää henkilökohtaista lääketiedettä. Toisaalta se on monogeeninen sairaus, jonka aiheuttavat tietyn geenin mutaatiot. Kokonaisuuden patofysiologia on hyvin tunnettu ja terapeuttiset kohteet ovat selkeät. Lisäksi taudin diagnosointi edellyttää geneettistä testausta tautityypin tunnistamiseksi, joten tarkka geenivirhe määritetään kussakin tapauksessa.,10
Tällä hetkellä kaksi hyvin erilaista lähestymistapoja ovat suunnattu korjaaminen perus vika: gene therapy, joilla pyritään korjaamaan geneettinen muutos, ja molekyyli hoitoa, joilla pyritään korjaamaan toiminnallinen vika proteiini-tasolla. Geeniterapian painopiste on normaalien geenikopioiden käyttöönotto CF-potilaiden hengitysteissä. Siihen lisätään rekombinantti virusvektori, jonka DNA on uutettu ja korvattu uudella terapeuttisella DNA: lla. Virusvektori toimii välineenä uuden DNA: n työntämiseksi kohdesoluun., Tähän mennessä on käytetty erilaisia viruksia, kuten adenovirusta tai lentivirusta. Lisäksi on kehitetty myös muita kuin virushiukkasia, kuten DNA: n asettamiseen kykeneviä nanohiukkasia.11 tähänastiset tulokset ovat kuitenkin olleet huonoja, sillä esitellyn geenin ilmentymisen kesto oli lyhyt molemmilla vektorityypeillä.12 UK Gene Therapy Konsortio kehittää kliinisen vaiheen II arvioida kliinistä tehokkuutta optimoitu plasmidic/liposomaalista DNA-vektori.13 rekrytointitavoite on 130 potilasta, ja tuloksia odotetaan vuonna 2014 (NCT01621867).,14
toisaalta CFTR-proteiinin toiminnan palauttamiseen tähtäävä hoito on onnistunut paremmin. Viime vuosina tulokset alkavat tulla läpi lääkkeistä, jotka vaikuttavat suoraan CFTR-proteiiniin. Itse asiassa tammikuussa 2012 ensimmäinen lääke Gly551Asp-mutaatiovirheiden korjaamiseen markkinoitiin Yhdysvalloissa. Seuraavissa kappaleissa aiomme tarkistaa saatavilla olevat tiedot edistymisestä henkilökohtaisen lääketieteen CF ja saatavilla hoitoja, joilla pyritään korjaamaan vika aiheuttaa taudin proteiini tasolla., Tässä tarkastelussa käytetään nimikkeistöä, jota käytetään ihmisen Genomivaihteluyhteiskunnan kehittämien CFTR-geenimutaatioiden kuvauksessa15.
Mutaatioita ja Proteiinia Vika
CF on autosomaalinen resessiivinen perinnöllinen sairaus, joten mutaatio on oltava läsnä sekä kopiot CFTR geeni vaikuttaa. Tähän mennessä yli 1900 CFTR-geenin mutaatiot liittyvät tauti on tunnistettu koodaavan sekvenssin, messenger-RNA: ta tai muita elementtejä. CFTR-geenin mutaatiot ovat kuultavissa kystisen fibroosin Mutaatiotietokannassa.,16 ensimmäinen mutaatio on kuvattu, ja yleisin maailmanlaajuisesti, on Phe508del, mutta on olemassa muita erityisiä mutaatioita vaihtelevalla taajuudella eri etnisten ryhmien. Espanjassa, keskimääräinen taajuus Phe508del mutaatio on välillä 50% ja 60% kaikki opiskellut kromosomeja, toiseksi yleisin on Gly542X 4% -8%, jonka jälkeen Asn1303Lys 2% -4% tapauksista. Tähän mennessä kuvatut mutaatiot luokitellaan kuuteen tyyppiin tai luokkaan taudin aiheuttavan mekanismin mukaan.17 tällaisia mutaatioita on yhteenveto Kuvassa. 1., Luokka I mutaatiot johtavat ennenaikainen stop kodonissa messenger RNA, joka estää käännös täydellistä proteiinia. Näin tuotettu proteiini on lyhyt ja toimimaton. Luokan II mutaatioita koodata rakenteellisesti epänormaali ja väärin laskostuneet proteiini, joka on poistaa endoplasmakalvosto ennen solun pinnalla. CF: n yleisin mutaatio, Phe508del, kuuluu tähän ryhmään. Luokkien III-VI mutaatioissa proteiinit saavuttavat solun pinnan, mutta eivät toimi kunnolla. Luokan III mutaatiot aiheuttavat heikentyneen kanavaaktivaation, joten kanavat pysyvät suljettuina., Luokan IV mutaatiot aiheuttavat ionin konduktanssin vähenemisen kanavan kautta. Luokka V mutaatioita koodata pieniä proteiineja, mikä vähentää määrä CFTR solun pinnalle, niin että tietty toiminto tapahtuu, mutta pienemmällä tasolla. Lopuksi, luokka VI mutaatiot johtavat lyhennetty puoliintumisaika, koska proteiini epävakautta ja voi myös vahingoittaa asetuksen naapurimaiden CFTR-kanavia solun pinnalla.
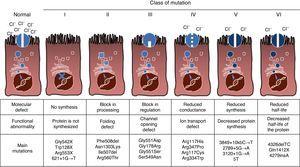
kystiseen fibroosiin liittyvien mutaatioiden tyypit.
CFTR Modulaattorit
Kolme luokkaa, on tunnistettu kehittämään lääkkeitä korjaamiseen CFTR-proteiinia.11 ensimmäinen ryhmä ovat ennenaikaiset codon suppressorit (luokan I mutaatiot). Nämä lääkkeet estävät tämän ennenaikaisen Stop-kodonin tunnistamisen, jotta proteiinisynteesi voi jatkua loppuun asti. Toinen ryhmä ovat CFTR correctors. Nämä yhdisteet ovat suunniteltu korjaamaan vikoja liikenteen taitettu proteiinia (luokka II mutaatioita) solukalvoon, jossa se voi toimia lähes normaalisti., Kolmas ryhmä koostuu niin sanotuista CFTR-potentiaattoreista. Nämä ovat lääkkeitä, jotka on suunniteltu kohdistamaan CFTR-proteiinia solun pinnalla sen toiminnan parantamiseksi. Näin nämä potentiaattorit voivat vaikuttaa luokan III, IV, V ja VI mutaatioihin. Tällä hetkellä, lukuisia molekyylejä käyttäen nämä eri mekanismit ovat tutkimuksen kohteena, joista yksi on jo saavuttanut markkinoilla: Ivacaftor (VX-770) on CFTR voimistava hyväksytty USA: ssa tammikuussa 2012 hoitoon CF-potilaat yli 6-vuotiaille, jotka ovat Gly551Asp mutaatio.,
luokan I Mutaatiohoidot
noin 10% CF-tapauksista johtuu luokan I mutaatioista. Ensimmäiset tähän luokkaan käytetyt lääkkeet olivat aminoglykosideja. Useita vuosia sitten gentamisiinilla kerrottiin olevan kyky peittää ennenaikainen stop-kodoni, joka estää CFTR-proteiinin synteesin. Tämä saavutetaan lisäämällä aminohappo, jonka avulla ribosomin jatkaa lukemista geeni, tuottaa kokoillan proteiinia. Prekliiniset tutkimukset osoittivat, että proteiinia voitiin syntetisoida hiirimallissa ja 35% proteiinin toiminnasta todettiin in vitro.,18,19 gentamisiinin laskimonsisäisen annostelun vaikutusta arvioitiin kahdessa tutkimuksessa CF-potilailla, joilla oli erityyppisiä I luokan mutaatioita. Yksi tehtiin Yhdysvalloissa viidellä potilaalla,20 ja toisessa 18 potilasta Ranskassa.21 vaikka vastaus oli myönteinen, tulokset vaihtelivat kuitenkin paljon, joten hyöty ei ollut universaali. Lisäksi toksisuusongelmat aminoglykosidien kanssa vaikuttivat epäedulliseen profiiliin.
synteettinen vaihtoehto on ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., Tämä on molekyyli, jonka avulla ribosomit voivat lukea geneettistä informaatiota ”ohittaen” ennenaikaisen stop-kodonin, tuottaen toimivan CFTR-proteiinin.Atalureenin farmakokinetiikka on osoitettu eläinmalleissa ja vaiheen II tutkimuksissa. Kaksi pientä alustavia tutkimuksia,23,24 ryhmä CF-potilailla, joita hoidettiin suun kautta ataluren osoitti parantaminen elektrofysiologiset häiriöt tauti, joka nostaa solujen määrä nenä ilmaista proteiinin pinnalla., Myöhemmin, toinen pieni tutkimus 19 potilasta, arvioitiin eri annoksia ataluren antaa suun kautta aina, 8h, kanssa parannuksia CFTR toimintaa ja kliiniset parametrit ja hyvä turvallisuusprofiili.25 tulokset vaiheen III kliinisessä tutkimuksessa ataluren, että ei ollut vielä virallisesti julkaistu raportoitiin vuonna 2012 Pohjois-Amerikan Kystinen Fibroosi Konferenssi. Yhteensä 238 yli 6-vuotiasta potilasta satunnaistettiin saamaan atalureenia 10, 10, 20 mg/kg tai lumelääkettä 8 tunnin välein 48 viikon ajan., Ei ollut merkittäviä eroja FEV1 vuonna ataluren lumelääkkeeseen verrattuna 48 viikon hoidon jälkeen (-2.5% ataluren vs -5.5% lumelääkeryhmässä, P=ns). Kun stratifioitu krooninen nebulized antibioottien käyttö oli 6,7% ero keskimääräinen muutos 48 viikon jälkeen hyväksi ataluren potilaille ei tobramysiini, kun ei ollut eroa muutos FEV1 saaneilla nebulized tobramysiini. Jotain vastaavaa tapahtui prosenttiosuus pahenemisvaiheita, jossa ero ryhmien välillä oli merkittävä., Kun kerrostunut potilaat ataluren ryhmä ei nebulized tobramysiini osoitti, että prosenttiosuus vähentää pahenemisvaiheiden 43% verrattuna lumeryhmään. Nenän potentiaalinen ero ja hikikloriditestit eivät osoittaneet eroa ryhmien välillä sumutettujen antibioottien käytöstä riippumatta. Kirjoittajat totesi, että hyödyt oli suurempi potilailla, jotka eivät saaneet krooninen antibioottihoidon kanssa nebulized aminoglykosidi, spekuloidaan, että tobramysiini ja ataluren vuorovaikutuksessa klo ribosomaalisen tasolla, tuottavat vastakkainasettelua, kun sitä käytetään samanaikaisesti.,26
Luokka II Mutaatio Hoidot
Luokka II-mutaatioita, on yleisin mutaatio tauti (Phe508del), ovat läsnä suuri määrä CF-potilailla, mikä tekee niistä ensisijainen tavoite CF-tutkimus. On tutkittu erilaisia molekyylejä, joista suurimman osan on kehittänyt Vertex Pharmaceuticals Inc. Ensimmäinen korjaaja yhdiste, lumacaftor (VX-809), osoitti hyvää in vitro tehoa, parantaa kloridi liikenne 14%.,27 Kuitenkin, tulokset olivat hieman pettymys potilaille, koska parannuksia kloridipitoisuus hiki oli hyvin pieni (7mmol/l) ja ei tapahtunut muutoksia nenän potentiaalia.28
vaikutukset CFTR voimistava ivacaftor (VX-770), huumeiden luokka III-mutaatioita (ks. kuvaus alla), on myös tutkittu potilailla, jotka olivat homotsygoottisia Phe508del. Tulosten perusteella LÖYTÄÄ tutkimuksen osoittavat, että ivacaftor ei liity merkittävää paranemista FEV1-arvon, elämän laatu tai määrä pahenemisvaiheita lumelääkkeeseen verrattuna.,29
Koska lumacaftor voi auttaa toimituksen Phe508del-CFTR solun pinnalla ja ivacaftor lisää aukioloaikoja ja kloridi johtuminen kautta epiteelin solujen, parantaa taustalla Phe508del vika voi olla mahdollista, kun yhdistelmä molekyylejä. In vitro-tutkimusten yhdistettyjen ivacaftor ja lumacaftor hengitysteiden epiteelin kanssa Phe508del mutaatio on osoittanut, että lumacaftor lisää CFTR kloridi liikenne 15% omasta, ja kun ivacaftor on lisätty, liikenne kasvaa lähes 30%., Tätä lääkeyhdistelmää on tutkittu vaiheen II tutkimuksessa potilailla, joilla on Phe508del-mutaatio. Koko tuloksia ei ole vielä julkaistu, mutta alustavien tietojen mukaan suotuisa vaikutus keuhkojen toimintaa homotsygooteilla Phe508del, mutta ei heterotsygoottisuuden.30 Kaksi kliinisissä tutkimuksissa potilailla 12 vuotta tai vanhempi homotsygoottinen mutaatio Phe508del kehitetään arvioida yhdistetty ivacaftor ja lumacaftor; nämä ovat LIIKENNE (NCT01807923)31 ja LIIKENNE (NCT01807949)32 tutkimukset. Tulosten perusteella noin puolet CF-populaatiosta voi saada CFTR-moduloivaa hoitoa., Tutkimuksia tarvitaan kuitenkin yhdistelmähoidon vaikutuksen arvioimiseksi Phe508del-heterotsygoottisilla yksilöillä.
Toinen vaihtoehto on, että korjaaja yhdiste VX-661, ja tutkimukset ovat parhaillaan käynnissä. Sen tehokkuutta testataan yksin ja yhdessä ivakaftorin (VX-770) kanssa, ja tulokset ovat pian saatavilla (NCT01531673).33
Luokka III Mutaatio Hoidot
Ivacaftor (VX-770) on CFTR voimistava, joka säätelee toimintaa epänormaalia proteiinia.,34 Tämä molekyyli on alun perin suunniteltu parantamaan CFTR-toiminto kulttuurien hengitysteiden epiteelin soluja kuljettaa yhden Gly551Asp mutaatio.34 se on ensimmäinen Yhdysvalloissa ja Euroopassa hyväksytty lääke CF: n hoitoon potilailla, joilla on Gly551Asp-mutaatio. Kun kapasiteetti ivacaftor parantaa kloridin kulkeutumista solukalvon osoitettiin in vitro, ensimmäisen vaiheen II kokeet suoritettiin 39 potilasta.35 tässä tutkimuksessa CFTR-proteiinin toiminta parani kolmen päivän kuluttua hoidon aloittamisesta, saavuttaa kloridipitoisuudet hiki jopa normaalille tasolle., Näiden tulosten perusteella seurasi kaksi muuta kliinistä tutkimusta: STRIVE-tutkimus 144: llä vähintään 12-vuotiaalla POTILAALLA36 ja ENVISION-tutkimus, jossa oli mukana 52 6-11-vuotiasta lasta.37 Molemmissa tutkimuksissa oli mukana potilaita, joilla vähintään yksi Gly551Asp mutaatio ja FEV1-arvon välillä 40% ja 105%, jotka olivat aluksi seurasi ajaksi 14 päivää ja sitten satunnaistetusti 150mg suun ivacaftor tai lumelääkettä kahdesti päivässä ajaksi 48 viikkoa., Suoritettuaan 48 viikon hoidon aikana potilaille annettiin mahdollisuus jatkaa pitkittäinen avoimessa tutkimuksessa, JATKUVAT tutkimuksessa (NCT01117012),38 96 viikkoa.
PYRIMME potilaat ivacaftor ryhmä oli parannus 10,6% FEV1 (ensisijainen päätetapahtuma) alkaa päivästä 15 hoidon, joka säilyi aikana 48 viikkoa kestäneessä tutkimuksessa. Lisäksi lasku kloridipitoisuus hiki oli havaittu (keskiarvo: -48.7 mmol/L), parantunut elämänlaatu, 55% vähentää pahenemisvaiheita ja painonnousu 2,7 kg.,
tulokset KUVITELLA tutkimus vastaa pitkälti niitä tutkimuksen nuorilla ja aikuisilla, sillä erotuksella, että elämänlaatu ei saavuttanut tilastollista eroa. Haitalliset vaikutukset useammin havaittu hoitoryhmässä sekä PYRKIÄ ja KUVITELLA tutkimukset olivat ylempien hengitysteiden infektiot, nenän tukkoisuus, kurkkukipu, huimaus ja ihottuma., Alustavat tulokset ensimmäisten 12 viikon jälkeen jatkuvat tutkimukset osoittavat, että keuhkojen toiminta (FEV1), hengitysoireet ja painonnousu ivakaftorilla hoidetuilla potilailla säilyy tänä aikana. Lisäksi potilaiden alaryhmässä, joka siirtyi lumelääkettä ivacaftor lähtötilanteessa JATKUVAT kokenut 10.8% parannus FEV1-arvon vähintään 15 päivää, ja 13% 12 viikossa, yhdessä vähentää pahenemisvaiheiden.,39
Huolimatta hyviä tuloksia ivacaftor hoidossa Gly551Asp mutaatio lapset yli 6 vuotta ja aikuisille 48 viikkoa, joitakin kysymyksiä on edelleen ratkaisematta. Ensinnäkin lääkettä ei ole testattu alle 6-vuotiailla lapsilla. Näyttää kuitenkin järkevältä korjata vika ennen peruuttamatonta vahinkoa tapahtuu, ottaen huomioon, että keuhkojen osallistuminen alkaa ennen kuuden vuoden ikää. Tällä ikähaarukalla on meneillään tutkimus (NCT01705145).40 toinen vaihtoehto olisi kokeilla myös muita III luokan mutaatioita., Tältä osin yhdeksää muuta mutaatiota koskevat in vitro-tutkimukset ovat osoittaneet hyvin samanlaisia tuloksia, 41 joten on kohtuullista odottaa samanlaisia tuloksia potilailla. Meneillään on faasin III kliininen tutkimus yli 6-vuotiailla potilailla, joilla on muita luokan III mutaatioita (KONTINUE-ja KONNEKTIOTUTKIMUKSET; NCT01614470).42,43 Kolmanneksi, vaikka mitään hoitoa on saatavilla muun mutaatio luokat (IV–VI), CFTR potentiaattoreina voi olla yhtä hyödyllistä näissä tapauksissa., Tässä yhteydessä tehdään kliininen tutkimus, jossa arvioidaan ivakaftorin tehoa Arg117His – luokan IV mutaatiossa (KONDUKTITUTKIMUS; NCT01614457).44 ivakaftorin tehoa ja pitkäaikaisturvallisuutta 48 viikon jälkeen ei ole vielä varmistettu. Se G551D Observational Study (TAVOITE; NCT01521338)45 havaintoihin perustuva tutkimus, seuraavat potilaat yli 6-vuotiaat saavat ivacaftor., Se on suunnattu raportointi tehoa ja turvallisuutta pitkän aikavälin ivacaftor, yhdessä muiden tuloksia edun mukaista, että ovat tulehdusvälittäjäaineiden yskös, mukosiliaarisen puhdistumaan, ja ruoansulatuskanavan pH. Tulokset ovat odotettavissa vuoden 2013 lopulla.
päätelmät
CF on esimerkki sairaudesta, jolla on hyvät mahdollisuudet hyödyntää henkilökohtaista lääketiedettä. Tällä hetkellä kaksi hyvin erilaista lähestymistapaa tavoitteena on korjata perus vika: gene therapy, joilla pyritään korjaamaan geneettinen muutos, ja hoidon molekyylejä, joiden tarkoituksena on korjata toiminnallisia vika proteiini-tasolla., Jälkimmäinen on alkanut näyttää lupaavia tuloksia eri molekyylien kehitystä, ja yksi heistä (ivacaftor) on jo kaupan Gly551Asp luokan III mutaatio, erinomaisia tuloksia lapsilla yli 6 vuotta, nuorille ja aikuisille. Lopullisena tavoitteena on tarjota oikaisu – ja potentiaattorihoitoja kaikille CF-potilaille riippumatta heidän mutaatiostaan. Koska tulokset näiden ja muiden uusien molekyylien näy, on todennäköistä, että tietty molekyyli tai yhdistelmä tarvitaan kullekin potilaalle, riippuen nykyisiä mutaatioita., Joka tapauksessa, tulevaisuus on lupaava, ja on otettu tärkeitä askelia kohti saada hoitoa, joka tulee tehokkaasti toimia tämän sairauden syy.
eturistiriidat
kirjoittajat eivät ilmoita eturistiriitoja.