Indledning
færdiggørelsen af det humane genom-projekt var en relevant milepæl for medicinsk viden, der giver de oplysninger, der er nødvendige for at forstå de unikke karakteristika for hver enkelt.1 den logiske konsekvens af denne viden ville være at være i stand til at anvende specifikke diagnostiske test og behandlinger til hver patient baseret på deres individuelle genetiske information. Denne nye form for lægehjælp kaldes personlig medicin.,2 på trods af de store fremskridt, der har ført til viden om det menneskelige genom, har dets oversættelse til diagnostiske og personaliserede behandlinger været mindre end forventet. På nuværende tidspunkt er der skridt i denne retning med to store initiativer: systembiologi3 og farmakogenetik.,4 det endelige mål med disse initiativer er at udvikle en medicinsk praksis tilpasset hver enkelt persons egenskaber, der kan forudsige begyndelsen eller forløbet af en bestemt sygdom, tillade passende forebyggelsesstrategier at blive etableret og endelig gøre det muligt for patienten at deltage i beslutningsprocessen. Dette er blevet kaldt P4 medicin.5
cystisk fibrose (CF) er fortsat den mest almindelige og dødelige genetiske sygdom blandt kaukasiere., Det sker med en hastighed på 1 i 2500-6000 nyfødte, afhængigt af region og etnisk oprindelse, og i en del af raske bærere, varierende mellem 1:20 og 37.6 I Spanien, takket være den gradvise indførelse af neonatal screening programmer i de forskellige samfund, en lavere forekomst af CF er nu ved at blive set, end det tidligere var skønnet i 2009, dvs, 1/4430 levendefødte i Galicien, 1/4339 i Castile-Leon, 1/5376 i Murcia, 1/5840 i Catalonien, og 1/6602 i de Baleariske Øer.7 Det anslås, at der er 70000 CF-patienter over hele verden.,8 sygdommen er forårsaget af mutationer i genet, der koder for det regulatoriske protein cystisk fibrose transmembranledningsregulator (CFTR), en chloridkanal involveret i frigivelsen af adenosintrifosfat og regulering af andre iontransportkanaler. Dette protein er udtrykt i respiratoriske epitelceller, bugspytkirtel, galdeveje, svedkirtler og genitourinært system., Ændring fører til en abnormitet i ion-transport, så patienterne producerer tyk, klæbrig slim, der træsko kanalerne af orgel, hvor det er placeret, og så den ændring præsenterer multisystemic effekter, der bestemmer bred vifte af kliniske manifestationer af CF. På trods af store fremskridt i behandlingen af CF, der har resulteret i længere overlevelse (nuværende median skønnes på 37,5 år),9 der er stadig en lang vej at gå for at sikre, at patienter med CF har en kvantitet og kvalitet af liv svarer til, at personer uden sygdommen., I denne sammenhæng er nye behandlinger for at mindske sygeligheden og øge overlevelsen nødvendige.
CF er et eksempel på en sygdom, der er godt positioneret til at drage fordel af personlig medicin. På den ene side er det en monogen sygdom forårsaget af mutationerne i et specifikt gen. Enhedens patofysiologi er godt karakteriseret, og de terapeutiske mål er klare. Endvidere kræver diagnose af sygdommen genetisk test til identifikation af sygdomstypen, så den nøjagtige genetiske defekt bestemmes i hvert tilfælde.,10
i øjeblikket er to meget forskellige tilgange rettet mod at korrigere den grundlæggende defekt: genterapi, der sigter mod at korrigere den genetiske ændring, og molekyleterapi, der sigter mod at korrigere den funktionelle defekt på proteinniveauet. Fokus for genterapi er introduktionen af normale genkopier i luftvejene hos CF-patienter. Det indebærer indsættelse af en rekombinant viral vektor, hvis DNA er blevet ekstraheret og erstattet af det nye terapeutiske DNA. Den virale vektor tjener som et køretøj til indsættelse af det nye DNA i målcellen., Forskellige typer vira, såsom adenovirus eller lentivirus, er blevet brugt til dato. Desuden er der også udviklet ikke-virale partikler, såsom nanopartikler, der er i stand til at indsætte DNA.11 imidlertid har resultaterne hidtil været dårlige, fordi varigheden af ekspression af det introducerede gen var kort med begge typer vektorer.12 Det britiske Genterapikonsortium udvikler et klinisk fase II-forsøg til evaluering af den kliniske effektivitet af en optimeret plasmid/liposomal DNA-vektor.13 rekrutteringsmålet er 130 patienter, og der forventes resultater i 2014 (NCT01621867).,14
på den anden side har terapi rettet mod at genoprette funktionen af CFTR-proteinet været mere vellykket. I de senere år er resultaterne begyndt at komme igennem på lægemidler, der virker direkte på CFTR-proteinet. Faktisk blev det første lægemiddel til korrektion af Gly551Asp-mutationsdefekter i januar 2012 markedsført i USA. I de følgende afsnit vil vi gennemgå de tilgængelige oplysninger om udviklingen af personlig medicin til CF og tilgængelige behandlinger med det formål at korrigere defekten, der forårsager sygdommen på proteinniveau., I denne gennemgang vil den nomenklatur, der anvendes til beskrivelse af CFTR-genmutationer udviklet af det humane genom Variation Society15, blive anvendt.
mutationer og Proteindefekt
CF er en autosomal recessiv arvelig sygdom, så mutationen skal være til stede i begge kopier af CFTR-genet for at blive påvirket. Til dato er over 1900 CFTR-genmutationer forbundet med sygdommen blevet identificeret i kodningssekvensen, messenger RNA eller andre elementer. Mutationer i CFTR-genet er tilgængelige til konsultation i databasen for cystisk fibrose-Mutation.,16 den første mutation beskrevet, og den mest almindelige på verdensplan, er Phe508del, men der er andre specifikke mutationer med varierende frekvens blandt forskellige etniske grupper. I Spanien, den gennemsnitlige frekvens af Phe508del mutation er mellem 50% og 60% af alle de undersøgte kromosomer, den anden mest hyppige er Gly542X med 4%-8%, efterfulgt af Asn1303Lys i 2%-4% af tilfældene. De hidtil beskrevne mutationer klassificeres i seks typer eller klasser i henhold til mekanismen, der forårsager sygdommen.17 disse typer mutationer er opsummeret i Fig. 1., Klasse i-mutationer fører til et for tidligt stopkodon i messenger RNA, som forhindrer oversættelse af det komplette protein. Således er det producerede protein kort og ikke-fungerende. Klasse II-mutationer koder for et strukturelt unormalt og forkert foldet protein, der fjernes af det endoplasmatiske retikulum, inden de når celleoverfladen. Den mest almindelige mutation i CF, Phe508del, tilhører denne gruppe. I tilfælde af mutationer i klasse III til vi når proteiner celleoverfladen, men fungerer ikke korrekt. Klasse III mutationer forårsager en nedsat kanalaktivering, så kanaler forbliver lukkede., Klasse IV mutationer forårsager et fald i ionledningsevne gennem kanalen. Klasse V-mutationer koder for mindre proteiner, hvilket resulterer i en reduceret mængde CFTR i celleoverfladen, så en bestemt funktion forekommer, men på et reduceret niveau. Endelig fører klasse vi-mutationer til en forkortet halveringstid på grund af proteinstabilitet og kan også skade reguleringen af nærliggende CFTR-kanaler i celleoverfladen.
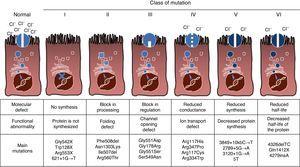
typer af mutationer i cystisk fibrose.
CFTR Modulatorer
Tre vigtigste klasser er blevet identificeret i udviklingen af lægemidler til reparation af CFTR-protein.11 Den første gruppe er for tidlige stopkodonundertrykkere (klasse i-mutationer). Disse lægemidler forhindrer identifikation af dette for tidlige stopkodon, så proteinsyntesen kan fortsætte indtil færdiggørelsen. Den anden gruppe er CFTR-korrektorerne. Disse forbindelser er designet til at korrigere defekter i transporten af foldet protein (klasse II-mutationer) til cellemembranen, hvor det muligvis kan fungere næsten normalt., Den tredje gruppe består af de såkaldte CFTR potentiatorer. Disse er lægemidler designet til at målrette CFTR-proteinet på celleoverfladen for at forbedre dets funktion. Således kan disse potentiatorer virke på klasse III, IV, V og vi mutationer. I øjeblikket undersøges adskillige molekyler, der bruger disse forskellige mekanismer, hvoraf den ene allerede har nået markedet: Ivacaftor (v.-770) er en CFTR-potentiator, der blev godkendt i USA i januar 2012 til behandling af CF-patienter over 6 år, der har Gly551Asp-mutationen.,10% af CF-tilfælde er forårsaget af klasse i-mutationer. De første lægemidler, der blev brugt til denne klasse, var aminoglycosider. For flere år siden blev gentamicin rapporteret at have evnen til at maskere det for tidlige stopkodon, der forhindrer syntese af CFTR-proteinet. Dette opnås ved indsættelse af en aminosyre, der gør det muligt for ribosomet at fortsætte med at læse genet og producere et protein i fuld længde. Prækliniske undersøgelser viste, at proteinet kunne syntetiseres i en musemodel, og 35% af proteinfunktionen blev genvundet in vitro.,18,19 effekten af intravenøs administration af gentamicin blev vurderet i to undersøgelser af CF-patienter med forskellige typer af klasse i-mutationer. Den ene blev udført i USA hos fem patienter,20 og en anden omfattede 18 patienter i Frankrig.21 selvom svaret var positivt, varierede resultaterne meget, så fordelen var ikke universel. Desuden bidrog toksicitetsproblemer med aminoglycosider til en ugunstig profil.
et syntetisk alternativ er ataluren (ptc124; PTC Therapeutics, South Plainfield, NJ, USA)., Dette er et molekyle designet til at gøre det muligt for ribosomer at læse den genetiske information, mens de “springer over” det for tidlige stopkodon og producerer et funktionelt CFTR-protein.22 atalurens farmakokinetik er påvist i dyremodeller og i fase II-forsøg. I to små foreløbige undersøgelser viste 23, 24 En gruppe CF-patienter behandlet med oral ataluren forbedring af sygdommens elektrofysiologiske abnormiteter med en stigning i antallet af celler i næsen, der udtrykker proteinet på deres overflade., Senere, en anden lille undersøgelse af 19 patienter evalueret forskellige doser af ataluren administreres oralt hver 8h, med forbedringer i CFTR aktivitet og kliniske parametre og en god sikkerhedsprofil.25 resultaterne af et klinisk fase III-forsøg med ataluren, der endnu ikke var formelt offentliggjort, blev rapporteret i 2012 North American Cystic fibrose Conference. I alt 238 patienter over 6 år blev randomiseret til at modtage ataluren 10, 10, 20 mg/kg eller placebo hver 8.time i 48 uger., Der var ingen signifikante forskelle i FEV1 i ataluren versus placebo efter 48 ugers behandling (-2.5% ataluren vs -5.5% placebo, P=ns). Når stratificeret af kronisk nebulized brug af antibiotika, var der en 6.7% forskel i gennemsnitlig ændring efter 48 uger til fordel for ataluren for patienter, som ikke på tobramycin, mens der var ingen forskel i ændring i FEV1 i dem, der modtager nebulized tobramycin. Noget lignende skete med procentdelen af eksacerbationer, hvor forskellen mellem begge grupper var signifikant., Ved stratificering viste patienterne i ataluren-gruppen ikke på forstøvet tobramycin et procentvis fald i eksacerbationer på 43% sammenlignet med placebogruppen. Nasal potentialforskel og svedchloridtest viste ingen forskel mellem grupper, uanset brugen af forstøvede antibiotika. Forfatterne konkluderede, at fordelene var større hos patienter, som ikke modtager kronisk antibiotisk behandling med nebulized aminoglycosid, at spekulere i, at tobramycin og ataluren kommunikerede på et ribosom niveau, der producerer antagonisme er, når de anvendes samtidig.,26
klasse II-Mutationsbehandlinger
klasse II-mutationer, der er den hyppigste mutation af sygdommen (Phe508del), er til stede hos et stort antal CF-patienter, hvilket gør dem til et primært mål i CF-forskning. Forskellige molekyler er blevet undersøgt, hvoraf de fleste er udviklet af Verte.Pharmaceuticals Inc. Den første korrektorforbindelse, lumacaftor (V.-809), viste God in vitro-effekt, hvilket forbedrede kloridtransport i 14%.,27 resultaterne var imidlertid noget skuffende hos patienter, fordi forbedringerne i chloridkoncentration i sved var meget små (7 mmol/l), og der var ingen ændringer i nasal potentiale.28
virkningerne af CFTR stimulator ivacaftor (VX-770), et lægemiddel for klasse III-mutationer (se beskrivelse nedenfor), er også blevet undersøgt hos patienter, der var homozygote for Phe508del. Resultaterne af DISCOVER-undersøgelsen viser, at ivacaftor ikke er forbundet med en signifikant forbedring i FEV1, livskvalitet eller antallet af eksacerbationer versus placebo.,29
Da lumacaftor kan hjælpe levering af Phe508del-CFTR celleoverfladen og ivacaftor øger åbningstid og chlorid varmeledning gennem epithelial cell, forbedring af den underliggende Phe508del defekt kan være muligt med kombinationen af de to molekyler. In vitro-undersøgelser af kombineret ivacaftor og lumacaftor i luftvejene epithelia med Phe508del mutation har vist, at lumacaftor øger CFTR klorid-transport med 15% om sin egen, og når ivacaftor er tilføjet, transport stiger til næsten 30%., Denne kombination af lægemidler er undersøgt i en fase II-undersøgelse hos patienter med Phe508del-mutation. De fulde resultater er endnu ikke offentliggjort, men de oprindelige data antyder en gavnlig effekt på lungefunktionen i homo .ygot Phe508del, men ikke i Hetero .ygositet.30 To kliniske forsøg i patienter, der er 12 år eller ældre homozygot for mutationen Phe508del er ved at blive udviklet til at vurdere, kombineret ivacaftor og lumacaftor; disse er TRAFIKKEN (NCT01807923)31 og TRANSPORT (NCT01807949)32 undersøgelser. Resultaterne kan give omkring halvdelen af CF-befolkningen mulighed for at modtage CFTR-modulerende terapi., Imidlertid er undersøgelser nødvendige for at evaluere effekten af kombinationsbehandling hos Phe508del Hetero .ygote individer.
et andet alternativ er corrector compound V.-661, og undersøgelser er i øjeblikket i gang. Dens effektivitet testes alene og i kombination med ivacaftor (V.-770), og resultaterne vil snart være tilgængelige (NCT01531673).33
klasse III Mutationsbehandlinger
Ivacaftor (v.-770) er en CFTR-potentiator, der modulerer funktionen af det unormale protein.,34 dette molekyle blev oprindeligt designet til at forbedre CFTR-funktionen i kulturer af respiratoriske epitelceller, der bærer en enkelt Gly551Asp-mutation.34 Det er det første lægemiddel, der er godkendt i USA og Europa til behandling af CF hos patienter, der bærer gly551asp-mutationen. Efter at ivacaftors kapacitet til forbedring af kloridtransport gennem cellemembranen blev påvist in vitro, blev de første fase II-forsøg udført med 39 patienter.35 i denne undersøgelse forbedrede CFTR-proteinfunktionen tre dage efter behandlingsstart og nåede chloridkoncentrationer i sved på op til normale niveauer., Med disse resultater fulgte to yderligere kliniske undersøgelser: STRIVE-undersøgelse hos 144 patienter i alderen 12 år eller mere36 og ENVISION-undersøgelse inklusive 52 børn mellem 6 og 11 år.37 Begge undersøgelser indgår patienter med mindst en Gly551Asp mutation og FEV1 mellem 40% og 105% der blev i første omgang fulgt i en periode på 14 dage og derefter randomiseret til at modtage 150mg af oral ivacaftor eller placebo to gange om dagen i en periode på 48 uger., Efter at have afsluttet 48 ugers behandling fik patienterne mulighed for at fortsætte i en langsgående åben undersøgelse, PERSIST-undersøgelsen (NCT01117012),38 i 96 uger.
I STRIVE havde patienter i ivacaftor-gruppen en forbedring på 10, 6% i FEV1 (primært endepunkt) fra dag 15 af behandlingen, som blev opretholdt i løbet af 48-ugers undersøgelsen. Desuden kan et fald i den chlorid koncentration i sved blev observeret (betyder: -48.7 mmol/L), med forbedret livskvalitet, 55% reduktion i antallet af eksacerbationer og en vægt-gevinst på 2,7 kg.,
resultaterne af ENVISION-undersøgelsen falder stort set sammen med resultaterne af undersøgelsen hos unge og voksne med den forskel, at livskvaliteten ikke nåede statistisk forskel. De bivirkninger, der hyppigere blev observeret i behandlingsgruppen i både STRIVE-og ENVISION-undersøgelser, var øvre luftvejsinfektioner, næseoverbelastning, ondt i halsen, svimmelhed og hududslæt., De foreløbige resultater efter de første 12 uger af PERSIST-undersøgelsen afslører, at forbedringer i lungefunktion (FEV1), åndedrætssymptomer og vægtøgning blandt patienter, der behandles med ivacaftor, opretholdes i denne periode. Desuden, den undergruppe af patienter, der skiftede fra placebo til at ivacaftor ved baseline i FORTSÆTTER oplevet en 10.8% forbedring i FEV1 på 15 dage, og 13% på 12 uger, sammen med en reduktion i tilbagefald.,39
På trods af de gode resultater med ivacaftor i behandlingen af Gly551Asp-mutationen hos børn over 6 år og voksne efter 48 uger, er der stadig nogle problemer, der skal løses. For det første er stoffet ikke blevet testet hos børn under 6 år. Det forekommer imidlertid fornuftigt at rette fejlen, før der opstår irreversibel skade, i betragtning af at lungeinddragelse begynder før seks år. Et forsøg i denne aldersgruppe er i øjeblikket i gang (NCT01705145).40 for det andet ville et andet alternativ være at også prøve andre klasse III-mutationer., I denne henseende har in vitro-undersøgelser af ni andre mutationer vist meget lignende resultater,41, så det er rimeligt at forvente lignende resultater hos patienter. Der er et igangværende klinisk fase III-forsøg med patienter over 6 år med andre klasse III-mutationer (KONTINUE-og KONNEKTIONSSTUDIER; NCT01614470).42,43 for det tredje, selvom der ikke er nogen behandling tilgængelig for resten af mutationsklasserne (IV–VI), kan CFTR-potentiatorer være lige så gavnlige i disse tilfælde., Der udføres et klinisk forsøg i denne henseende, der vurderer effektiviteten af ivacaftor i Arg117His klasse IV-mutationen (KONDUCT-undersøgelse; NCT01614457).44 endelig er effektivitet og langsigtet sikkerhed for ivacaftor ud over 48 uger endnu ikke fastlagt. G551D-Observationsundersøgelsen (mål; NCT01521338)45 er en observationsundersøgelse efter patienter ældre end 6 år, der fik ivacaftor., Det sigter mod at rapportere effektiviteten og sikkerheden ved langvarig ivacaftor sammen med andre resultater af interesse, der inkluderer inflammatoriske mediatorer i sputum, mucociliær clearance og gastrointestinal pH. resultaterne forventes i slutningen af 2013.
konklusioner
CF er et eksempel på en sygdom, der er godt positioneret til at drage fordel af personlig medicin. På nuværende tidspunkt sigter to meget forskellige tilgange til at korrigere den grundlæggende defekt: genterapi med det formål at korrigere den genetiske ændring og terapi med molekyler med det formål at korrigere den funktionelle defekt på proteinniveauet., Sidstnævnte begynder at vise lovende resultater for forskellige molekyler under udvikling, og en af dem (ivacaftor) er allerede markedsført til Gly551Asp klasse III mutation, med fremragende resultater hos børn over 6 år, unge og voksne. Det endelige mål er at give korrektions-og potentiatorbehandlinger til alle CF-patienter uanset deres mutation. Da resultaterne af disse og andre nye molekyler vises, er det sandsynligt, at der er behov for et specifikt molekyle eller en kombination for hver patient, afhængigt af deres eksisterende mutationer., Under alle omstændigheder er fremtiden lovende, og der er taget vigtige skridt i retning af at opnå en behandling, der effektivt vil virke på årsagen til denne sygdom.
interessekonflikter
forfatterne erklærer ingen interessekonflikter.