Úvod
dokončení projektu lidského genomu bylo relevantním milníkem pro lékařské znalosti, poskytuje informace nezbytné pro pochopení jedinečné vlastnosti každého jednotlivce.1 logickým důsledkem těchto znalostí by bylo, aby bylo možné aplikovat specifické diagnostické testy a léčby pro každého pacienta na základě jejich individuálních genetických informací. Tato nová forma lékařské péče se nazývá personalizovaná medicína.,2 navzdory velkému pokroku, který vedl k poznání lidského genomu, byl jeho překlad do diagnostické a personalizované léčby menší, než se očekávalo. V současné době probíhají kroky tímto směrem se dvěma hlavními iniciativami: systémová biologi3 a farmakogenetika.,4 konečným cílem těchto iniciativ je vypracovat lékařskou praxi, přizpůsobené vlastnosti každého jednotlivce, který může předvídat nástup nebo průběh určitého onemocnění, umožňují vhodných preventivních strategií a konečně, aby pacient podílet na rozhodování. Tomu se říká lék P4.5
cystická fibróza (CF) zůstává nejčastějším a smrtelným genetickým onemocněním mezi bělochy., To se vyskytuje v poměru 1 2500-6000 novorozenců, v závislosti na regionu a etnického původu, a to v poměru zdravých nosičů, pohybuje mezi 1:20 a 37.6, Ve Španělsku, a to díky postupné zavedení novorozenecké screeningové programy v různých komunitách, nižší výskyt CF je nyní vidět, než se dříve odhadovalo v roce 2009, tj. 1/4430 živě narozených dětí v Haliči, 1/4339 v Kastilie-Leon, 1/5376 v Murcia, 1/5840 v Katalánsku, a 1/6602 na Baleárských Ostrovech.7 odhaduje se, že na celém světě trpí 70000 CF.,8 nemoc je způsobena mutací v genu kódující regulační protein cystická fibróza transmembránový vodivost regulátor (CFTR), chloridový kanál podílí na uvolňování adenosintrifosfátu a nařízení jiných transport iontů kanály. Tento protein je exprimován v respiračních epiteliálních buňkách, pankreatu, žlučových cestách, potních žlázách a genitourinárním systému., Jeho změna vede k abnormality v transportu iontů, tak že pacienti produkují hustý, lepkavý hlen, který ucpává vývodů orgánu, kde se nachází, a to tak, že se změna představuje multisystémové účinky, které určují širokou škálu klinických projevů CF. I přes významné pokroky v léčbě CF, které mají za následek delší přežití (medián se odhaduje na 37,5 let),9 tam je ještě dlouhá cesta, aby zajistily, že pacienti s CF mají množství a kvalitu života podobný jedinců bez onemocnění., V této souvislosti jsou nezbytné nové léčby ke snížení morbidity a zvýšení přežití.
CF je příkladem onemocnění, které má dobrou pozici k využití personalizované medicíny. Na jedné straně je to monogenní onemocnění způsobené mutacemi v konkrétním genu. Patofyziologie entity je dobře charakterizována a terapeutické cíle jsou jasné. Kromě toho diagnóza onemocnění vyžaduje genetické testování pro identifikaci typu onemocnění, takže přesná genetická vada je stanovena v každém případě.,10.
V současné době, dva velmi odlišné přístupy jsou zaměřeny na korekci základní vady: genová terapie, zaměřené na korekci na genetické změny, a molekula terapie, zaměřené na korekci funkční vada na proteinové úrovni. Zaměření genové terapie je zavedení normálních genových kopií do dýchacích cest pacientů s CF. Zahrnuje vložení rekombinantního virového vektoru, jehož DNA byla extrahována a nahrazena novou terapeutickou DNA. Virový vektor slouží jako prostředek pro vložení nové DNA do cílové buňky., Dosud byly použity různé typy virů, jako je adenovirus nebo lentivirus. Kromě toho byly také vyvinuty nevirové částice, jako jsou nanočástice schopné vkládat DNA.11 dosavadní výsledky však byly špatné, protože doba exprese zavedeného genu byla u obou typů vektorů krátká.12 konsorcium pro genovou terapii ve Velké Británii vyvíjí klinickou studii fáze II, která vyhodnotí klinickou účinnost optimalizovaného vektoru plazmidické / liposomální DNA.13 cílem náboru je 130 pacientů a výsledky se očekávají v roce 2014 (NCT01621867).,14
na druhé straně byla terapie zaměřená na obnovení funkce proteinu CFTR úspěšnější. V posledních letech se začínají objevovat výsledky u léků, které působí přímo na protein CFTR. Ve skutečnosti byl v lednu 2012 uveden na trh první lék na opravu defektů mutace Gly551Asp v USA. V následujících částech přezkoumáme dostupné informace o pokroku personalizované medicíny pro CF a dostupné léčby zaměřené na nápravu vady způsobující onemocnění na úrovni bílkovin., V tomto přehledu bude použita nomenklatura použitá pro popis mutací genů CFTR vyvinutých společností Human Genome Variation Society15.
Mutace a Bílkovin Vady
CF je autosomálně recesivní dědičné onemocnění, takže mutace musí být přítomen v obou kopií CFTR genu být ovlivněna. K dnešnímu dni bylo v kódovací sekvenci identifikováno více než 1900 genových mutací CFTR spojených s onemocněním, messengerovou RNA nebo jinými prvky. Mutace v genu CFTR jsou k dispozici ke konzultaci v databázi mutací cystické fibrózy.,16 první popsaná mutace a nejběžnější na celém světě je Phe508del, ale existují i jiné specifické mutace s různou frekvencí mezi různými etnickými skupinami. Ve Španělsku, průměrná frekvence Phe508del mutace je mezi 50% a 60% všech studovaných chromozomů, druhou nejčastější je Gly542X s 4%-8%, následuje Asn1303Lys v 2%-4% případů. Dosud popsané mutace jsou rozděleny do šesti typů nebo tříd podle mechanismu způsobujícího onemocnění.17 tyto typy mutací jsou shrnuty na obr. 1., Mutace třídy I vedou k předčasnému zastavení kodonu v messenger RNA, což zabraňuje překladu úplného proteinu. Produkovaný protein je tedy krátký a nefunkční. Mutace třídy II kódují strukturálně abnormální a špatně složený protein, který je odstraněn endoplazmatickým retikulem před dosažením buněčného povrchu. Nejběžnější mutace v CF, Phe508del, patří do této skupiny. V případě mutací ve třídách III až VI se proteiny dostanou na buněčný povrch, ale nefungují správně. Mutace třídy III způsobují sníženou aktivaci kanálu, takže kanály zůstávají zavřené., Mutace třídy IV způsobují pokles iontové vodivosti kanálem. Mutace třídy v kódují menší proteiny, což vede ke snížení množství CFTR v buněčném povrchu, takže dochází k určité funkci, ale na snížené úrovni. Nakonec mutace třídy VI vedou ke zkrácenému poločasu v důsledku nestability bílkovin a mohou také poškodit regulaci sousedních CFTR kanálů v buněčném povrchu.
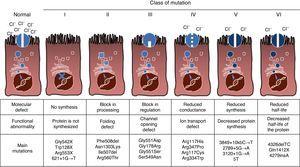
typy mutací u cystické fibrózy.
CFTR Modulátory
Tři hlavní třídy byly identifikovány ve vývoji léků pro opravy CFTR protein.11 první skupinou jsou předčasné Stop codon supresory (mutace třídy I). Tyto léky zabraňují identifikaci tohoto předčasného Stop kodonu, takže syntéza bílkovin může pokračovat až do dokončení. Druhou skupinou jsou korektory CFTR. Tyto sloučeniny jsou určeny k nápravě defektů v transportu složeného proteinu (mutace třídy II) na buněčnou membránu, kde může fungovat téměř normálně., Třetí skupinu tvoří tzv. CFTR potenciátory. Jedná se o léky určené k cílení proteinu CFTR na buněčný povrch, aby se zlepšila jeho funkce. Tyto potenciátory tak mohou působit na mutace třídy III, IV, V A VI. V současné době, četné molekuly pomocí těchto různých mechanismů jsou předmětem šetření, z nichž jeden dosáhl již na trhu: Ivacaftor (VX-770) je potenciátor CFTR schválen v USA v lednu 2012 pro léčbu CF pacientů starších než 6 let věku, kteří mají Gly551Asp mutace.,
léčba mutací třídy I
přibližně 10% případů CF je způsobeno mutacemi třídy I. První léky používané pro tuto třídu byly aminoglykosidy. Před několika lety bylo hlášeno, že gentamicin má schopnost maskovat předčasný stop kodon zabraňující syntéze proteinu CFTR. Toho je dosaženo vložením aminokyseliny, která umožňuje ribozomu pokračovat ve čtení genu a produkovat protein v plné délce. Preklinické studie prokázaly, že protein může být syntetizován v myším modelu a 35% proteinové funkce bylo získáno in vitro.,18,19 účinek intravenózního podání gentamicinu byl hodnocen ve dvou studiích u pacientů s CF s různými typy mutací třídy I. Jeden byl proveden v USA u pěti pacientů, 20 a další zahrnoval 18 pacientů ve Francii.21 ačkoli byla odpověď pozitivní, výsledky se značně lišily, takže přínos nebyl univerzální. Navíc Problémy s toxicitou aminoglykosidy přispěly k nepříznivému profilu.
syntetickou alternativou je ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., To je molekula, navržen tak, aby ribozomy na čtení genetické informace, zatímco „přeskakování“ předčasný stop kodon, výrobu funkčního CFTR protein.22 farmakokinetika atalurenu byla prokázána u zvířecích modelů a ve studiích fáze II. Ve dvou malých předběžné studie,23,24 skupina CF pacientů léčených perorálními ataluren ukázal zlepšení elektrofyziologické abnormality nemoci, s nárůstem počtu buněk v nose vyjadřující bílkovin na jejich povrch., Následně, další malé studii 19 pacientů hodnoceny různé dávky ataluren podáván perorálně každých 8h, se zlepšením v CFTR činnosti a klinických parametrů a dobrý bezpečnostní profil.25 výsledky klinické studie fáze III atalurenu, která dosud nebyla formálně zveřejněna, byly hlášeny na konferenci o severoamerické cystické fibróze v roce 2012. Celkem 238 pacientů starších než 6 let bylo randomizováno ataluren 10, 10, 20 mg/kg nebo placebo každé 8h po dobu 48 týdnů., Nebyly žádné významné rozdíly v FEV1 v ataluren versus placebo po dobu 48 týdnů léčby (-2.5% ataluren vs až 5,5% u placeba, P=ns). Když v třídění podle chronická nebulizovaný používání antibiotik, tam byl o 6,7% rozdíl v průměrná změna po 48 týdnech ve prospěch ataluren u pacientů, u na tobramycin, zatímco tam byl žádný rozdíl ve změně v FEV1 u pacientů léčených nebulizovaný tobramycin. Něco podobného se stalo s procentem exacerbací, kde byl rozdíl mezi oběma skupinami významný., Když vrstevnatý, pacienti v ataluren skupina není na nebulizovaný tobramycin ukázaly procentuální pokles exacerbací o 43% ve srovnání s placebem. Nosní potenciální rozdíl a testy chloridu potu neprokázaly žádný rozdíl mezi skupinami, bez ohledu na použití nebulizovaných antibiotik. Autoři dospěli k závěru, že přínosy jsou vyšší u pacientů podstupujících chronickou léčbu antibiotiky s nebulizovaný aminoglykosidová, spekulovat, že tobramycin a ataluren styku na ribozomální úrovni, produkovat antagonismus při současném použití.,26
Class II Mutace Ošetření
II. Třída mutací, nejčastější mutace onemocnění (Phe508del), jsou přítomny ve velkém počtu CF pacientů, což je primární cíl v CF výzkumu. Byly studovány různé molekuly, z nichž většina byla vyvinuta společností Vertex Pharmaceuticals Inc. První korektorová sloučenina, lumacaftor (VX-809), vykazovala dobrou účinnost in vitro a zlepšila transport chloridu ve 14%.,27 výsledky však byly u pacientů poněkud zklamáním, protože zlepšení koncentrace chloridu v potu bylo velmi malé (7mmol/l) a nedošlo ke změnám v nosním potenciálu.28
účinky CFTR potenciátor ivacaftor (VX-770), lék pro třídy III mutace (viz popis níže), byly také zkoumány u pacientů, kteří byli homozygotní pro Phe508del. Výsledky studie DISCOVER ukazují, že ivacaftor není spojen s významným zlepšením FEV1, kvality života nebo počtu exacerbací oproti placebu.,29.
Od lumacaftor může napomáhat dodání Phe508del-CFTR na povrchu buněk a ivacaftor zvyšuje otevírací doba a chlorid vedení přes epiteliálních buněk, zlepšení podkladových Phe508del vada může být možné s kombinací obou molekul. In vitro studie kombinované ivacaftor a lumacaftor v respiračním epitelu s Phe508del mutace ukázaly, že lumacaftor zvyšuje CFTR chlorid dopravy o 15%, na jeho vlastní, a když ivacaftor je přidáno, doprava se zvyšuje na téměř 30%., Tato kombinace léčiv byla zkoumána ve studii fáze II u pacientů s mutací Phe508del. Úplné výsledky dosud nebyly zveřejněny, ale počáteční údaje naznačují příznivý účinek na funkci plic u homozygotního Phe508delu, ale ne v heterozygozitě.30 Dvou klinických studiích u pacientů od 12 let nebo starší homozygotní pro mutaci Phe508del jsou vyvíjeny s cílem vyhodnotit v kombinaci ivacaftor a lumacaftor; tyto jsou DOPRAVNÍ (NCT01807923)31 a DOPRAVY (NCT01807949)32 studií. Výsledky by mohly umožnit přibližně polovině populace CF, aby dostávala modulační terapii CFTR., K vyhodnocení účinku kombinované terapie u heterozygotních jedinců Phe508del jsou však zapotřebí studie.
další alternativou je korektorová sloučenina VX-661 a v současné době probíhají studie. Jeho účinnost je testována samostatně, tak v kombinaci s ivacaftor (VX-770), a výsledky budou brzy k dispozici (NCT01531673).33
léčba mutací třídy III
Ivacaftor(VX-770) je potenciátor CFTR, který moduluje funkci abnormálního proteinu.,34 tato molekula byla původně navržena pro zvýšení funkce CFTR v kulturách buněk respiračního epitelu nesoucích jedinou mutaci Gly551Asp.34 jedná se o první lék schválený v USA a Evropě pro léčbu CF u pacientů nesoucích mutaci Gly551Asp. Poté, co byla in vitro prokázána kapacita ivacaftoru pro zlepšení transportu chloridů přes buněčnou membránu, byly provedeny první studie fáze II s 39 pacienty.35 v této studii se funkce proteinu CFTR zlepšila tři dny po zahájení léčby a dosáhla koncentrací chloridu v potu až do normálních hladin., S těmito výsledky následovaly dvě další klinické studie: studie STRIVE, u 144 pacientů ve věku 12 let nebo více36 a studie ENVISION včetně 52 dětí ve věku od 6 do 11 let.37 Obě studie zahrnovaly pacienty s alespoň jedním Gly551Asp mutace a FEV1 mezi 40% a 105%, které byly původně sledovala po dobu 14 dní a poté randomizováni a užívali 150 mg ústní ivacaftor nebo placeba dvakrát denně po dobu 48 týdnů., Po dokončení 48 týdnů léčby dostali pacienti možnost pokračovat v podélné otevřené studii, ve studii PERSIST (NCT01117012), 38 po dobu 96 týdnů.
ve skupině STRIVE měli pacienti ve skupině ivacaftor zlepšení o 10, 6% u FEV1 (primární koncový bod) od 15.dne léčby, které bylo zachováno během 48týdenní studie. Kromě toho, snížení koncentrace chloridu v potu bylo pozorováno (průměr: -48.7 mmol/L), s lepší kvalitou života, 55% snížení exacerbací a přibývání na váze 2,7 kg.,
výsledky PŘEDSTAVIT studie do značné míry shodují s těmi, studie u dospívajících a dospělých, s tím rozdílem, že kvalita života nedosáhl statistické rozdíl. Nežádoucí účinky častěji pozorované v léčené skupině ve studiích STRIVE a ENVISION byly infekce horních cest dýchacích, nazální kongesce, bolest v krku, závratě a kožní vyrážka., Předběžné výsledky po prvních 12 týdnů PŘETRVÁVAT studie ukazují, že zlepšení funkce plic (FEV1), respirační příznaky, a přibývání na váze u pacientů léčených s ivacaftor jsou udržována během tohoto období. Navíc, v podskupině pacientů, kteří přešli z placeba na ivacaftor na výchozí hodnoty PŘETRVÁVAJÍ zkušený 10,8% zlepšení v FEV1 na 15 dnů, a 13% na 12 týdnů, spolu s snížení exacerbací.,39
navzdory dobrým výsledkům přípravku ivacaftor při léčbě mutace Gly551Asp u dětí starších 6 let a dospělých ve věku 48 týdnů je třeba vyřešit některé problémy. Za prvé, lék nebyl testován u dětí mladších 6 let. Zdá se však rozumné opravit vadu dříve, než dojde k nevratnému poškození, vzhledem k tomu, že postižení plic začíná před dosažením věku šesti let. V současné době probíhá studie v tomto věkovém rozmezí (NCT01705145).40 za druhé, další alternativou by bylo také vyzkoušet jiné mutace třídy III., V tomto ohledu studie in vitro na devíti dalších mutacích prokázaly velmi podobné výsledky, 41 proto je rozumné očekávat podobné výsledky u pacientů. Probíhá klinická studie fáze III u pacientů starších 6 let s jinými mutacemi třídy III (studie KONTINUE a KONNECTION; NCT01614470).42,43 Zatřetí, ačkoli není k dispozici žádná léčba pro ostatní třídy mutací (IV–VI), potenciátory CFTR mohou být v těchto případech stejně prospěšné., V tomto ohledu se provádí klinická studie, která hodnotí účinnost ivacaftoru v mutaci Arg117His třídy IV (studie KONDUCT; nct01614457).44 účinnost a dlouhodobá bezpečnost přípravku ivacaftor po 48 týdnech ještě nebyla stanovena. Na G551D Observační Studie (CÍL; NCT01521338)45 je observační studie těchto pacientů starších než 6 let léčených ivacaftor., Je zaměřena na podávání zpráv o účinnosti a bezpečnosti dlouhodobé ivacaftor, spolu s dalšími výsledky zájmů, které patří zánětlivých mediátorů ve sputu, očista sliznic odbavení, a gastrointestinální pH. Výsledky jsou očekávány na konci roku 2013.
závěry
CF je příkladem onemocnění, které má dobrou pozici k využití personalizované medicíny. V současné době, dva velmi odlišné přístupy, cílem je, jaká základní vady: genová terapie zaměřené na korekci na genetické změny, a terapie se molekuly zaměřené na korekci funkční vada na proteinové úrovni., Ta se začíná projevovat slibné výsledky pro různé molekuly ve vývoji, a jeden z nich (ivacaftor) je již na trhu pro Gly551Asp mutaci třídy III, s výbornými výsledky u dětí starších než 6 let, dospívajících a dospělých. Konečným cílem je poskytnout léčbu korektor a potenciátor pro všechny pacienty CF bez ohledu na jejich mutaci. Jak se objevují výsledky těchto a dalších nových molekul, je pravděpodobné, že pro každého pacienta bude zapotřebí specifická molekula nebo kombinace v závislosti na jejich stávajících mutacích., V každém případě je budoucnost slibná a byly podniknuty důležité kroky k získání léčby, která bude účinně působit na příčinu této nemoci.
střety zájmů
autoři nehlásí žádný střet zájmů.