Introduction
Der Abschluss des Human genome Project war ein wichtiger Meilenstein für medizinisches Wissen und lieferte die notwendigen Informationen, um die einzigartigen Eigenschaften jedes Einzelnen zu verstehen.1 Die logische Konsequenz dieses Wissens wäre, auf der Grundlage ihrer individuellen genetischen Information spezifische diagnostische Tests und Behandlungen auf jeden Patienten anwenden zu können. Diese neue Form der medizinischen Versorgung wird personalisierte Medizin genannt.,2 Trotz der großen Fortschritte, die zur Kenntnis des menschlichen Genoms geführt haben, war die Übersetzung in diagnostische und personalisierte Behandlungen jedoch geringer als erwartet. Gegenwärtig werden Schritte in diese Richtung mit zwei Hauptinitiativen unternommen: Systembiologie3 und Pharmakogenetik.,4 Das ultimative Ziel dieser Initiativen ist es, eine auf die Merkmale jedes Einzelnen zugeschnittene medizinische Praxis zu entwickeln, die den Ausbruch oder Verlauf einer bestimmten Krankheit vorhersagen, geeignete Präventionsstrategien festlegen und schließlich dem Patienten die Teilnahme an der Entscheidungsfindung ermöglichen kann. Dies wurde P4-Medizin genannt.5
Mukoviszidose (CF) bleibt die häufigste und tödlichste genetische Erkrankung bei Kaukasiern., Je nach Region und ethnischer Herkunft und in einem Anteil gesunder Träger, der zwischen 1:20 und 37, 6 in Spanien variiert, tritt sie mit einer Rate von 1 zu 2500-6000 Neugeborenen auf.Dank der fortschreitenden Einführung von Neugeborenen-Screening-Programmen in den verschiedenen Gemeinden ist jetzt eine geringere Inzidenz von CF zu beobachten als zuvor in 2009 geschätzt, d. H. 1/4430 Lebendgeburten in Galizien, 1/4339 in Kastilien-Leon, 1/5376 in Murcia, 1/5840 in Katalonien und 1/6602 auf den Balearen.7 Es wird geschätzt, dass es weltweit 70000 CF-Betroffene gibt.,8 Die Krankheit wird durch Mutationen in dem Gen verursacht, das für das regulatorische Protein Cystic Fibrosis Transmembran Conductance Regulator (CFTR) kodiert, einem Chloridkanal, der an der Freisetzung von Adenosintriphosphat und der Regulierung anderer Ionentransportkanäle beteiligt ist. Dieses Protein wird in respiratorischen Epithelzellen, Bauchspeicheldrüse, Gallenwege, Schweißdrüsen und Urogenitalsystem exprimiert., Seine Veränderung führt zu einer Abnormalität des Ionentransports, so dass Patienten dicken, klebrigen Schleim produzieren, der die Gänge des Organs verstopft, in dem er sich befindet, und so zeigt die Veränderung multisystemische Wirkungen, die das breite Spektrum klinischer Manifestationen von CF bestimmen. Trotz großer Fortschritte bei der Behandlung von CF,die zu einem längeren Überleben geführt haben (der gegenwärtige Median wird auf 37, 5 Jahre geschätzt) 9, ist es noch ein weiter Weg, um sicherzustellen, dass Patienten mit CF eine ähnliche Quantität und Lebensqualität haben wie Patienten ohne die Krankheit., In diesem Zusammenhang sind neue Behandlungen erforderlich, um die Morbidität zu verringern und das Überleben zu erhöhen.
CF ist ein Beispiel für eine Krankheit, die gut positioniert ist, um die personalisierte Medizin zu nutzen. Einerseits ist es eine monogene Erkrankung, die durch Mutationen in einem bestimmten Gen verursacht wird. Die Pathophysiologie der Entität ist gut charakterisiert, und die therapeutischen Ziele sind klar. Darüber hinaus erfordert die Diagnose der Krankheit Gentests zur Identifizierung des Krankheitstyps, so dass jeweils der genaue genetische Defekt bestimmt wird.,10
Gegenwärtig zielen zwei sehr unterschiedliche Ansätze auf die Korrektur des Grunddefekts ab: die Gentherapie zur Korrektur der genetischen Veränderung und die Molekültherapie zur Korrektur des funktionellen Defekts auf Proteinebene. Der Schwerpunkt der Gentherapie liegt auf der Einführung normaler Genkopien in die Atemwege von CF-Patienten. Es beinhaltet die Insertion eines rekombinanten viralen Vektors, dessen DNA extrahiert und durch die neue therapeutische DNA ersetzt wurde. Der virale Vektor dient als Vehikel zum Einfügen der neuen DNA in die Zielzelle., Bisher wurden verschiedene Arten von Viren wie Adenovirus oder Lentivirus verwendet. Darüber hinaus wurden auch nicht-virale Partikel, wie Nanopartikel, die DNA einfügen können, entwickelt.11 Die bisherigen Ergebnisse waren jedoch schlecht, da die Expressionsdauer des eingeführten Gens bei beiden Arten von Vektoren kurz war.12 Das britische Gentherapie-Konsortium entwickelt eine klinische Phase-II-Studie zur Bewertung der klinischen Wirksamkeit eines optimierten plasmidischen/liposomalen DNA-Vektors.13 Das Rekrutierungsziel ist 130 Patienten und Ergebnisse werden im Jahr 2014 erwartet (NCT01621867).,14
Andererseits war die Therapie zur Wiederherstellung der Funktion des CFTR-Proteins erfolgreicher. In den letzten Jahren beginnen die Ergebnisse bei Arzneimitteln, die direkt auf das CFTR-Protein einwirken. Tatsächlich wurde im Januar 2012 das erste Medikament zur Korrektur von Gly551Asp-Mutationsfehlern in den USA vermarktet. In den folgenden Abschnitten werden wir die verfügbaren Informationen über den Fortschritt der personalisierten Medizin für CF und die verfügbaren Behandlungen zur Korrektur des Defekts, der die Krankheit auf Proteinebene verursacht, überprüfen., In dieser Übersicht wird die Nomenklatur verwendet, die für die Beschreibung von CFTR-Genmutationen verwendet wird, die von der Human Genome Variation Society15 entwickelt wurden.
Mutationen und Proteindefekt
CF ist eine autosomal-rezessive Erbkrankheit, daher muss die Mutation in beiden Kopien des CFTR-Gens vorhanden sein, um betroffen zu sein. Bis heute wurden über 1900 mit der Krankheit assoziierte CFTR-Genmutationen in der kodierenden Sequenz, Messenger-RNA oder anderen Elementen identifiziert. Mutationen im CFTR-Gen können in der Mukoviszidose-Mutationsdatenbank eingesehen werden.,16 Die erste beschriebene und weltweit häufigste Mutation ist Phe508del, aber es gibt andere spezifische Mutationen mit unterschiedlicher Häufigkeit zwischen verschiedenen ethnischen Gruppen. In Spanien liegt die durchschnittliche Häufigkeit der Phe508del-Mutation zwischen 50% und 60% aller untersuchten Chromosomen, die zweithäufigste ist Gly542X mit 4% -8%, gefolgt von Asn1303Lys in 2% -4% der Fälle. Die bisher beschriebenen Mutationen werden je nach krankheitsverursachendem Mechanismus in sechs Typen oder Klassen eingeteilt.17 Diese Arten von Mutationen sind in Abb. 1., Klasse – I-Mutationen führen zu einem vorzeitigen Stopp-Codon in der Messenger-RNA, das die Translation des vollständigen Proteins verhindert. Somit ist das produzierte Protein kurz und nicht funktionsfähig. Mutationen der Klasse II kodieren für ein strukturell abnormes und fehlgefaltetes Protein, das vom endoplasmatischen Retikulum entfernt wird, bevor es die Zelloberfläche erreicht. Die häufigste mutation im CF, Phe508del, gehört zu dieser Gruppe. Bei Mutationen der Klassen III bis VI erreichen Proteine die Zelloberfläche, funktionieren aber nicht richtig. Mutationen der Klasse III verursachen eine verminderte Kanalaktivierung, sodass Kanäle geschlossen bleiben., Mutationen der Klasse IV verursachen eine Abnahme der Ionenleitfähigkeit durch den Kanal. Klasse-V-Mutationen kodieren kleinere Proteine, was zu einer reduzierten Menge an CFTR in der Zelloberfläche führt, so dass eine bestimmte Funktion auftritt, jedoch auf einem reduzierten Niveau. Schließlich führen Mutationen der Klasse VI aufgrund von Proteininstabilität zu einer verkürzten Halbwertszeit und können auch die Regulation benachbarter CFTR-Kanäle in der Zelloberfläche schädigen.
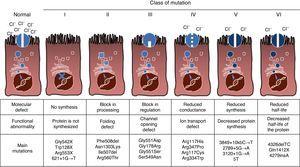
Arten von Mutationen bei Mukoviszidose.
CFTR-Modulatoren
Bei der Entwicklung von Arzneimitteln zur Reparatur von CFTR-Proteinen wurden drei Hauptklassen identifiziert.11 Die erste Gruppe sind vorzeitige Stop-Codon-Suppressoren (Mutationen der Klasse I). Diese Medikamente verhindern die Identifizierung dieses vorzeitigen Stop-Codons, so dass die Proteinsynthese bis zum Abschluss fortgesetzt werden kann. Die zweite Gruppe sind die CFTR-Korrektoren. Diese Verbindungen wurden entwickelt, um Defekte beim Transport von gefaltetem Protein (Mutationen der Klasse II) zur Zellmembran zu korrigieren, wo es möglicherweise fast normal funktionieren kann., Die dritte Gruppe besteht aus den sogenannten CFTR-Potentiatoren. Dies sind Medikamente, die entwickelt wurden, um das CFTR-Protein auf der Zelloberfläche anzuvisieren, um seine Funktion zu verbessern. Somit können diese Potentiatoren auf Mutationen der Klassen III, IV, V und VI einwirken. Derzeit werden zahlreiche Moleküle untersucht, die diese verschiedenen Mechanismen verwenden, von denen eines bereits auf dem Markt ist: Ivacaftor (VX-770) ist ein CFTR-Potentiator, der im Januar 2012 in den USA für die Behandlung von CF-Patienten über 6 Jahren mit der Gly551Asp-Mutation zugelassen wurde.,
Klasse-I-Mutationsbehandlungen
Etwa 10% der CF-Fälle werden durch Klasse-I-Mutationen verursacht. Die ersten Medikamente, die für diese Klasse verwendet wurden, waren Aminoglykoside. Vor einigen Jahren wurde berichtet, dass Gentamicin die Fähigkeit hat, die vorzeitige Stopp-Codon-Verhinderung der Synthese des CFTR-Proteins zu maskieren. Dies wird durch die Insertion einer Aminosäure erreicht, die es dem Ribosom ermöglicht, das Gen weiter zu lesen und ein Protein in voller Länge zu produzieren. Präklinische Studien zeigten, dass das Protein in einem Mausmodell synthetisiert werden konnte und 35% der Proteinfunktion in vitro wiederhergestellt wurden.,18,19 Die Wirkung der intravenösen Verabreichung von Gentamicin wurde in zwei Studien mit CF-Patienten mit verschiedenen Arten von Mutationen der Klasse I untersucht. Eine wurde in den USA an fünf Patienten durchgeführt, 20 und eine weitere an 18 Patienten in Frankreich.21 Obwohl die Resonanz positiv war, waren die Ergebnisse sehr unterschiedlich, so dass der Nutzen nicht universell war. Darüber hinaus trugen Toxizitätsprobleme mit Aminoglykosiden zu einem ungünstigen Profil bei.
Eine synthetische alternative ist ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA)., Dies ist ein Molekül, das Ribosomen ermöglicht, die genetische Information zu lesen, während das vorzeitige Stop-Codon „übersprungen“ wird und ein funktionelles CFTR-Protein erzeugt wird.22 Die Pharmakokinetik von Ataluren wurde in Tiermodellen und in Phase-II-Studien nachgewiesen. In zwei kleinen Vorstudien, 23, 24, zeigte eine Gruppe von CF-Patienten, die mit oraler Atalurie behandelt wurden, eine Verbesserung der elektrophysiologischen Anomalien der Erkrankung, wobei die Anzahl der Zellen in der Nase zunahm, die das Protein auf ihrer Oberfläche exprimierten., Anschließend untersuchte eine weitere kleine Studie mit 19 Patienten verschiedene Dosen von Ataluren, die alle 8 Stunden oral verabreicht wurden, mit Verbesserungen der CFTR-Aktivität und der klinischen Parameter und einem guten Sicherheitsprofil.25 Die Ergebnisse einer klinischen Phase-III-Studie mit Ataluren, die noch nicht offiziell veröffentlicht wurde, wurden 2012 auf der North American Cystic Fibrosis Conference berichtet. Insgesamt 238 Patienten, die älter als 6 Jahre waren, erhielten 48 Wochen lang randomisiert ataluren 10, 10, 20 mg/kg oder Placebo alle 8 Stunden., Es gab keine signifikanten Unterschiede in FEV1 in ataluren im Vergleich zu placebo nach 48 Wochen der Behandlung (-2.5% ataluren vs -5.5% placebo, P=ns). Bei einer Schichtung durch chronisch vernebelte Antibiotikakonsum gab es einen Unterschied von 6,7% in der mittleren Veränderung nach 48 Wochen zugunsten von Ataluren für Patienten, die nicht mit Tobramycin behandelt wurden, während es keinen Unterschied in der Veränderung von FEV1 bei Patienten gab, die vernebeltes Tobramycin erhielten. Ähnliches trat bei dem Prozentsatz der Exazerbationen auf, bei dem der Unterschied zwischen beiden Gruppen signifikant war., Bei der Schichtung zeigten die Patienten in der Atalurengruppe, die nicht mit vernebeltem Tobramycin behandelt wurden, eine prozentuale Abnahme der Exazerbationen um 43% im Vergleich zur Placebogruppe. Nasenpotentialdifferenz – und Schweißchloridtests zeigten keinen Unterschied zwischen Gruppen, unabhängig von der Verwendung vernebelter Antibiotika. Die Autoren kamen zu dem Schluss, dass der Nutzen bei Patienten, die keine chronische Antibiotikatherapie mit vernebeltem Aminoglykosid erhielten, größer war, und spekulierten, dass Tobramycin und Atalur auf ribosomaler Ebene interagierten und bei gleichzeitiger Anwendung Antagonismus erzeugten.,26
Klasse – II-Mutation Behandlungen
Klasse-II-Mutationen, die häufigste Mutation der Krankheit (Phe508del), sind bei einer großen Anzahl von CF-Patienten vorhanden, was sie zu einem primären Ziel in der CF-Forschung macht. Es wurden verschiedene Moleküle untersucht, von denen die meisten von Vertex Pharmaceuticals Inc.entwickelt wurden. Die erste Korrektorverbindung, Lumacaftor (VX-809), zeigte eine gute In-vitro-Wirksamkeit und verbesserte den Chloridtransport in 14%.,27 Die Ergebnisse waren jedoch bei den Patienten etwas enttäuschend, da die Chloridkonzentration im Schweiß sehr gering war (7 mmol/l) und es keine Veränderungen im Nasenpotential gab.28
Die Wirkungen des CFTR-Potentiators ivacaftor (VX-770), eines Arzneimittels für Mutationen der Klasse III (siehe Beschreibung unten), wurden auch bei Patienten untersucht, die für Phe508del homozygot waren. Die Ergebnisse der DISCOVER-Studie zeigen, dass Ivacaftor nicht mit einer signifikanten Verbesserung von FEV1, Lebensqualität oder der Anzahl der Exazerbationen im Vergleich zu Placebo verbunden ist.,29
Da Lumacaftor die Abgabe von Phe508del-CFTR an die Zelloberfläche unterstützen kann und Ivacaftor die Öffnungszeit und die Chloridleitung durch die Epithelzelle erhöht, kann mit der Kombination beider Moleküle eine Verbesserung des zugrunde liegenden Phe508del-Defekts möglich sein. In-vitro-Studien von kombinierten Ivacaftor und Lumacaftor in respiratorischen Epithelien mit Phe508del-Mutation haben gezeigt, dass Lumacaftor den CFTR-Chloridtransport allein um 15% erhöht, und wenn Ivacaftor hinzugefügt wird, erhöht sich der Transport auf fast 30%., Diese Kombination von Arzneimitteln wurde in einer Phase-II-Studie bei Patienten mit Phe508del-Mutation untersucht. Vollständige Ergebnisse wurden noch nicht veröffentlicht, aber erste Daten deuten auf eine positive Wirkung auf die Lungenfunktion bei homozygotem Phe508del hin, jedoch nicht bei Heterozygot.30 Zwei klinische Studien an Patienten ab 12 Jahren, die homozygot für die Mutation Phe508del sind, werden entwickelt, um kombinierte ivacaftor und lumacaftor zu bewerten; Dies sind die TRAFFIC (NCT01807923)31 und TRANSPORT (NCT01807949)32 Studien. Die Ergebnisse könnten es etwa der Hälfte der CF-Population ermöglichen, eine CFTR-modulierende Therapie zu erhalten., Es sind jedoch Studien erforderlich, um die Wirkung einer Kombinationstherapie bei heterozygoten Personen mit Phe508del zu bewerten.
Eine weitere Alternative ist die Korrektorverbindung VX-661, und derzeit laufen Studien. Seine Wirksamkeit wird allein und in Kombination mit Ivacaftor (VX-770) getestet und die Ergebnisse werden in Kürze verfügbar sein (NCT01531673).33
Klasse-III-Mutationsbehandlungen
Ivacaftor (VX-770) ist ein CFTR-Potentiator, der die Funktion des abnormalen Proteins moduliert.,34 Dieses Molekül wurde ursprünglich entwickelt, um die CFTR-Funktion in Kulturen von respiratorischen Epithelzellen mit einer einzigen Gly551Asp-Mutation zu verbessern.34 Es ist das erste in den USA und Europa zugelassene Medikament zur Behandlung von CF bei Patienten mit der Gly551Asp-Mutation. Nachdem die Fähigkeit von ivacaftor, den Chloridtransport durch die Zellmembran zu verbessern, in vitro nachgewiesen wurde, wurden die ersten Phase-II-Studien mit 39 Patienten durchgeführt.35 In dieser Studie verbesserte sich die CFTR-Proteinfunktion drei Tage nach Beginn der Behandlung und erreichte Chloridkonzentrationen im Schweiß von bis zu normalen Spiegeln., Mit diesen Ergebnissen folgten zwei weitere klinische Studien: Die Studie mit 144 Patienten ab 12 Jahren und die ENVISION-Studie mit 52 Kindern zwischen 6 und 11 Jahren.37 Beide Studien umfassten Patienten mit mindestens einer Gly551Asp-Mutation und FEV1 zwischen 40% und 105%, die zunächst über einen Zeitraum von 14 Tagen verfolgt und dann randomisiert wurden, um 150 mg orales Ivacaftor oder Placebo zweimal täglich für einen Zeitraum von 48 Wochen zu erhalten., Nach Abschluss der 48-wöchigen Behandlung erhielten die Patienten die Möglichkeit, in einer offenen longitudinalen Studie,der PERSIST-Studie (NCT01117012), 38 für 96-Wochen fortzufahren.
Bei Patienten in der Ivacaftor-Gruppe zeigte sich ab Tag 15 der Behandlung eine Verbesserung von 10, 6% bei FEV1 (primärer Endpunkt), die während der 48-wöchigen Studie beibehalten wurde. Weiterhin wurde eine Abnahme der Chloridkonzentration im Schweiß beobachtet (Mittelwert: -48,7 mmol/l), mit verbesserter Lebensqualität, einer 55% igen Verringerung der Exazerbationen und einer Gewichtszunahme von 2,7 kg.,
Die Ergebnisse der ENVISION-Studie stimmen weitgehend mit denen der Studie bei Jugendlichen und Erwachsenen überein, mit dem Unterschied, dass die Lebensqualität keinen statistischen Unterschied erreichte. Die Nebenwirkungen, die in der Behandlungsgruppe sowohl in der Studie als auch in der Studie von ENVISION häufiger beobachtet wurden, waren Infektionen der oberen Atemwege, verstopfte Nase, Halsschmerzen, Schwindel und Hautausschlag., Die vorläufigen Ergebnisse nach den ersten 12 Wochen der PERSIST-Studie zeigen, dass Verbesserungen der Lungenfunktion (FEV1), der Atmungssymptome und der Gewichtszunahme bei Patienten, die mit Ivacaftor behandelt wurden, während dieses Zeitraums erhalten bleiben. Darüber hinaus zeigte die Untergruppe der Patienten, die zu Studienbeginn in PERSIST von Placebo auf Ivacaftor umgestellt hatten, eine 10,8% ige Verbesserung der FEV1 nach 15 Tagen und 13% nach 12 Wochen zusammen mit einer Verringerung der Exazerbationen.,39
Trotz der guten Ergebnisse mit Ivacaftor bei der Behandlung der Gly551Asp-Mutation bei Kindern über 6 Jahren und Erwachsenen nach 48 Wochen bleiben einige Probleme zu lösen. Erstens wurde das Medikament nicht bei Kindern unter 6 Jahren getestet. Es scheint jedoch sinnvoll, den Defekt zu korrigieren, bevor irreversible Schäden auftreten, wenn man bedenkt, dass die Lungenbeteiligung vor dem sechsten Lebensjahr beginnt. Eine Studie in dieser Altersgruppe ist derzeit im Gange (NCT01705145).40 Zweitens wäre eine andere Alternative, auch andere Mutationen der Klasse III auszuprobieren., In dieser Hinsicht haben In-vitro-Studien an neun anderen Mutationen sehr ähnliche Ergebnisse gezeigt, so dass es vernünftig ist, ähnliche Ergebnisse bei Patienten zu erwarten. Es gibt eine laufende klinische Phase-III-Studie bei Patienten über 6 Jahren mit anderen Mutationen der Klasse III (KONTINUE-und KONNEKTIONSSTUDIEN; NCT01614470).42,43 Drittens können CFTR–Potentiatoren in diesen Fällen gleichermaßen vorteilhaft sein, obwohl für die übrigen Mutationsklassen (IV-VI) keine Behandlung verfügbar ist., Diesbezüglich wird eine klinische Studie durchgeführt, in der die Wirksamkeit von Ivacaftor bei der Arg117His-Mutation der Klasse IV bewertet wird (TRADUCT-Studie; NCT01614457).44 Schließlich wurde die Wirksamkeit und Langzeitsicherheit von Ivacaftor über 48 Wochen hinaus noch nicht nachgewiesen. Die G551D Observational Study (GOAL; NCT01521338)45 ist eine Beobachtungsstudie an Patienten, die älter als 6 Jahre sind und ivacaftor erhalten., Es zielt darauf ab, die Wirksamkeit und Sicherheit von Langzeit-Ivacaftor zu berichten, zusammen mit anderen Ergebnissen von Interesse, die Entzündungsmediatoren im Sputum, mukoziliäre Clearance und Magen-Darm-pH umfassen.
Schlussfolgerungen
CF ist ein Beispiel für eine Krankheit, die gut positioniert ist, um die personalisierte Medizin zu nutzen. Gegenwärtig zielen zwei sehr unterschiedliche Ansätze darauf ab, den Grunddefekt zu korrigieren: Gentherapie zur Korrektur der genetischen Veränderung und Therapie mit Molekülen zur Korrektur des funktionellen Defekts auf Proteinebene., Letzteres zeigt vielversprechende Ergebnisse für verschiedene Moleküle in der Entwicklung, und eines davon (Ivacaftor) wird bereits für die Gly551Asp-Klasse-III-Mutation vermarktet, mit hervorragenden Ergebnissen bei Kindern über 6 Jahren, Jugendlichen und Erwachsenen. Das Endziel ist es, allen CF-Patienten unabhängig von ihrer Mutation korrektorische und potentiatorische Behandlungen anzubieten. Wenn die Ergebnisse dieser und anderer neuer Moleküle erscheinen, ist es wahrscheinlich, dass je nach vorhandenen Mutationen für jeden Patienten ein bestimmtes Molekül oder eine bestimmte Kombination benötigt wird., In jedem Fall ist die Zukunft vielversprechend, und es wurden wichtige Schritte unternommen, um eine Behandlung zu erhalten, die effektiv auf die Ursache dieser Krankheit einwirkt.
Interessenkonflikte
Die Autoren erklären keine Interessenkonflikte.